Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia

Introduction
X-linked hypophosphataemia (XLH) is a rare metabolic bone disorder caused by pathogenic variants in the PHEX (phosphate-regulating endopeptidase homologue X-linked) gene, which is mainly expressed in bone (osteoblasts and osteocytes) and teeth (odontoblasts and cementoblasts). The incidence of XLH is 3.9 per 100,000 live births and the prevalence ranges from 1.7 to 4.8 per 100,000 people1,2,3. The pathogenesis of XLH is complex and incompletely understood. Many features of the disease can be explained by increased secretion of the phosphaturic hormone fibroblast growth factor 23 (FGF23) from bone. Binding of FGF23 to the FGF receptor 1–alpha-Klotho co-receptor complex results in downregulation of the sodium-dependent phosphate transporters NPT2a (encoded by SLC34A1) and NPT2c (encoded by SLC34A3), a decrease in 1,25-dihydroxyvitamin D (1,25(OH)2D) synthesis and an increase in 1,25(OH)2D degradation in the proximal renal tubules, ultimately causing hypophosphataemia and hypovitaminosis D4. The clinical consequences of XLH can include rickets, osteomalacia, bone pain, leg deformities (Fig. 1), premature fusion of the cranial sutures and disproportionate short stature that usually develops during the first 2 years of life5. Patients also show hypomineralization of teeth and are prone to tooth abscesses and periodontitis6 (Fig. 2). Adult patients with XLH may show pseudofractures due to osteomalacia as well as osteoarthritis, enthesopathies, spinal stenosis, hearing loss, depression and reduced quality of life7. The pathogenesis of osteoarthritis and enthesopathies and to what extent these are sequelae of certain primary manifestations of XLH is poorly understood8,9.

The patients show disproportionate short stature with genu varum (bowed legs) or genu valgum (knock-knees). The radiographs reveal severe leg bowing, partial fraying and irregularity of the distal femoral and proximal tibial growth plates. Note the lack of bone resorption features. Reprinted from ref. 17, CC BY 4.0.

a, Oral clinical view of a 5-year-old male patient with X-linked hypophosphataemia (XLH) showing a spontaneous dental abscess on the right upper temporary central incisor. The tooth shows no discoloration or carious lesion and the child and his mother reported no history of trauma. b, Maxillo-facial cellulitis due to spontaneous necrosis of the left upper temporary canine in the same patient at the age of 7 years. c, Panoramic radiograph of the same patient at the age of 8 years showing mixed dentition with characteristic dental features of XLH, including a normal (slightly thin) enamel layer, a radiolucent dentin layer with enlarged pulp chambers and prominent pulp horns on both temporary and permanent teeth. d, Oral clinical view of a 49-year-old woman with XLH who was diagnosed at the age of 4 years. The patient was treated with oral phosphate supplements and active vitamin D during growth for 12 years before the treatment was stopped at the age of 16 years. This treatment was resumed for 4 years from the age of 40 years before being replaced with burosumab, which had been taken for 5 years. e, Panoramic radiograph of the same patient showing generalized horizontal alveolar bone loss and teeth treated endodontically owing to dental infections.
Historically, XLH was treated with frequent doses of oral phosphate supplements and active vitamin D, which is sometimes referred to as ‘conventional treatment’. As serum phosphate returns to the trough level 1–2 h after administration of phosphate supplements, frequent doses are required to prolong the duration of relatively high serum phosphate levels. In addition, active vitamin D is required to prevent secondary hyperparathyroidism induced by phosphate supplements, which further promotes renal phosphate wasting, and to improve remineralization of the skeleton by enhancing vitamin D-dependent intestinal calcium and phosphate absorption10,11. This treatment can ameliorate rickets and osteomalacia, limit dental abscess formation and partly restore growth in children with XLH5,6. However, adult height is reduced in the majority of patients with XLH despite this therapy6. The limited efficacy is at least partly because both phosphate and active vitamin D further stimulate FGF23 secretion, resulting in a vicious circle of renal phosphate wasting12. This therapy is also associated with adverse effects, including hyperparathyroidism and nephrocalcinosis, and is poorly tolerated owing to gastrointestinal adverse effects and the need for frequent dosing5,6,13,14,15,16. These aspects were extensively discussed in our 2019 clinical practice recommendations for the diagnosis and management of XLH17.
Since 2018, burosumab, a fully humanized antibody against FG23, has become available for the treatment of XLH in Europe, USA and many other countries18,19,20,21,22,23,24. In patients with XLH, treatment with burosumab results in dose-dependent improvements in renal phosphate reabsorption, serum phosphate levels and hypophosphataemia-associated complications, including rickets and osteomalacia25. As evidence for the efficacy and safety of burosumab in children and adults with XLH were limited in 2018, we could only provide preliminary recommendations on the use of this therapy in our 2019 guideline.
Here, we present updated clinical practice recommendations for the diagnosis and management of XLH to assist clinicians caring for paediatric and adult patients. We incorporate new evidence on the diagnosis and management of patients with XLH, most of which relates to burosumab treatment. As XLH is a rare disease, the number of studies that enable recommendations with a high level of evidence is low. For this reason, we provide expert opinion when the literature is limited and clearly indicate the level of evidence for each recommendation.
These recommendations are the result of a structured process that began in 2022 and involved a series of online conferences with a team of experts and patient representatives. They have been endorsed by the European Society for Paediatric Nephrology, the European Society for Paediatric Endocrinology, the European Society of Endocrinology, the European Renal Association, the European Reference Network on Rare Endocrine Conditions, the European Reference Network on Rare Bone Disorders, the European Reference Network on Rare Kidney Diseases, the International Osteoporosis Foundation (IOF) Skeletal Rare Diseases Working Group, the European Calcified Tissue Society, the European Paediatric Orthopaedic Society Study Group on Metabolic and Genetic Bone Disorders, the European Society of Craniofacial Surgery, the European Society for Paediatric Neurosurgery and the European Federation of Periodontology, and will be further revised and endorsed periodically.
Methods
We followed the same methodology as for our 2019 guideline17. In brief, a core leadership group and a voting panel were assembled. The core group comprised specialists from endocrinology (D.S., M.L.B., L.S., P.K., L.R., A.L. and W.H.), nephrology (D.H., F.E., J.B., D.B. G.A. and E.L.), orthopaedic surgery (D.E., P.W. and L.S.), rheumatology (K.B. and K.M.J.), dentistry (M.B.D. and C.C.) and neurosurgery (F.D.R.), and representatives from XLH patient organizations (P.H., M.K. and O.G.). The voting group included 38 members with expertise in XLH, including members of the supporting societies and networks. The voting group members were asked to provide a level of agreement to the recommendations on a five-point scale (strongly disagree, disagree, neither agree/disagree, agree, strongly agree) (Delphi method). Failing a 70% level of consensus, recommendations were modified after discussion in the core group and reviewed again by the voting panel until a consensus level of at least 70% was achieved.
We developed patient (or population) covered; intervention; comparator, outcomes questions26 that were addressed in the literature searches and represent the basis for the recommendations. The population covered included paediatric and adult patients with XLH. Treatment benefits were evaluated against the no-treatment option, the patient status at baseline (that is, before therapy) or treatment with oral phosphate and active vitamin D or with burosumab as the comparators. The following outcomes were assessed: diagnosis, follow-up and treatment with respect to bone and dental disease, growth, quality of life, treatment-associated or disease-associated complications, comorbidities and transition to adult care.
The PubMed database was searched until 30 September 2023 and all articles and reports were considered, including randomized controlled trials (RCTs), uncontrolled or observational studies, registries, summaries and case reports, restricted to human studies in English. The following key MeSH terms were used to identify suitable studies: X-linked hypophosphatemia; X-linked hypophosphatemic rickets; hypophosphatemic rickets; familial hypophosphatemic rickets; PHEX; osteomalacia; burosumab; phosphate; active vitamin D; calcitriol. A second PubMed database search was done on 24 October 2024 using the same MeSH terms to incorporate new evidence obtained during the guideline process. Recommendations were graded according to the American Academy of Paediatrics grading system to develop the recommendations27, as shown in our 2019 guideline17. The quality of evidence was graded as high (A), moderate (B), low (C), very low (D) or not applicable (X). The strength of each recommendation was graded as strong, moderate, weak or discretionary (when no recommendation could be made).
Although phosphorus rather than phosphate is measured in serum or, in some clinics, in plasma, we use the term phosphate instead of phosphorus and refer to phosphate serum concentrations for consistency throughout the manuscript. We do not use the term ‘conventional treatment’ to refer to oral phosphate and active vitamin D, as most paediatric patients with XLH in Europe are now treated with burosumab.
Diagnosis of XLH
The clinical, biochemical and radiological features that suggest a diagnosis of XLH were described in detail in our 2019 guideline17. Here, we provide updated recommendations and focus on discussion of new aspects (Box 1).
For the assessment of phosphate homeostasis in adults, a fasting blood draw and 24-h urine collection are recommended because serum phosphate concentration and urinary calcium excretion vary with food intake throughout the day. However, this approach is not practical in infants and children. In the Canadian Laboratory Initiative on Pediatric Reference Intervals project, which is currently the most widely used paediatric reference study for blood analytes, and the Hannover Reference values for Pediatrics study, which established Lambda-Mu-Sigma-based continuous reference percentiles for blood and urinary laboratory parameters of phosphate homeostasis in children, sample collections were performed throughout the day, regardless of meals28,29. Non-fasting serum phosphate levels seem to be appropriate to detect hypophosphataemia and monitor burosumab treatment in paediatric patients with XLH using these reference values. However, the interval between the blood draw and the last meal or burosumab injection should be considered, and, if in doubt, we suggest repeated measurements to determine trends over time. A random spot urine test with calculation of urinary calcium to creatinine ratio is appropriate to estimate calcium excretion in children30. However, in those with very low urinary creatinine levels (for example, owing to low muscle mass), 24-h urine collection is recommended. Updated reference values for the maximum rate of renal tubular reabsorption of phosphate normalized to the glomerular filtration rate (TmP/GFR), urinary calcium and phosphate to creatinine ratios in children29,31 and a web calculator for TmP/GFR and age-adjusted and, where appropriate, sex-adjusted z-scores for serum phosphate, TmP/GFR, urinary calcium and phosphate to creatinine ratios for patients aged 0 to 18 years are available29,32 (Box 2). Of note, TmP/GFR measurements using the formula provided by Brodehl et al. are reliable in the fasting and non-fasting state33.
The differential diagnosis of XLH is based on the mechanisms leading to hypophosphataemia — namely, high parathyroid hormone (PTH) activity (leading to calcipenic rickets), inadequate phosphate absorption from the gut or renal phosphate wasting (leading to phosphopenic rickets) (Fig. 3). Renal phosphate wasting may be due to genetic or acquired tubular defects or to high levels of circulating FGF23 (Supplementary Table 1). When possible, the diagnosis of XLH should be confirmed by genetic testing.
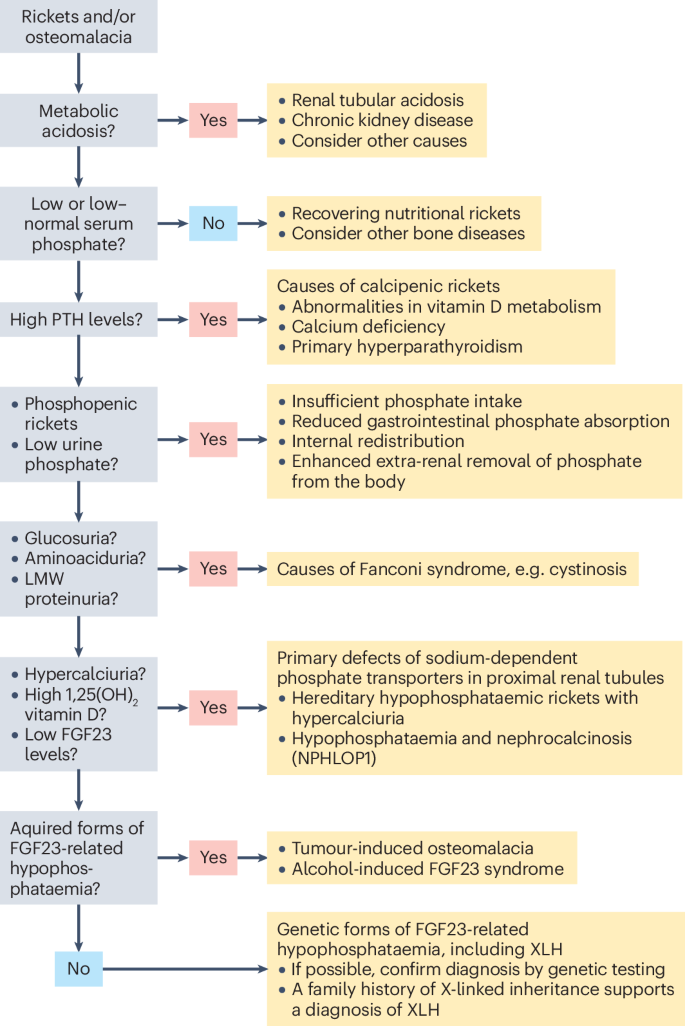
Patients usually present with rickets or osteomalacia and concomitant hypophosphataemia. The differential diagnosis is based on the mechanisms leading to hypophosphataemia, namely, high parathyroid hormone (PTH) activity (leading to calcipenic rickets or osteomalacia), inadequate phosphate absorption from the gut or renal phosphate wasting (leading to phosphopenic rickets or osteomalacia). A family history of X-linked inheritance with full penetrance in female carriers strongly supports the diagnosis of X-linked hypophosphataemia (XLH), which can be confirmed by genetic testing. FGF23, fibroblast growth factor 23; LMW, low molecular weight.
In the presence of hypophosphataemia, FGF23 synthesis and release from bone will decrease, resulting in plasma FG23 levels in the low or low–normal range2,34,35,36. In principle, FGF23 levels represent the key discriminant in non-calcipenic rickets, enabling FGF23-mediated inhibition of phosphate reabsorption by proximal tubular cells to be distinguished from primary renal tubular wasting, including various forms of Fanconi syndrome, including cystinosis prior to kidney failure (OMIM##219800), Dent disease (OMIM#300554, OMIM#300555), hypophosphataemia and nephrocalcinosis (OMIM#612286), reno-tubular syndrome 2 (OMIM#613388), and hereditary hypophosphataemic rickets with hypercalciuria (OMIM#241530)17 (Supplementary Table 1). These diseases are characterized by low or suppressed levels of FGF23.
If FGF23 levels are not suppressed, the differential diagnosis is limited to a few diseases, with XLH (OMIM#307800) being the most frequent, accounting for approximately 80% of cases17,37. Other potential diagnoses include autosomal-dominant hypophosphataemic rickets (OMIM#193100), autosomal-recessive hypophosphataemic rickets 1 (OMIM#241520); autosomal-recessive hypophosphataemic rickets 2 (OMIM#613312); Raine syndrome (OMIM#259775), fibrous dysplasia (OMIM#174800), tumour-induced osteomalacia, cutaneous skeletal hypophosphataemia syndrome (OMIM#163200) or epidermal nevus syndrome (OMIM#162900), osteoglophonic dysplasia (OMIM#166250), neurofibromatosis 1 (OMIM#162200) and hypophosphataemic rickets with hyperparathyroidism (OMIM#612089)17,37,38. Iron therapy with intravenous ferric carboxymaltose or iron isomaltoside may also promote high FGF23 levels. The cascade of biochemical changes that is induced by intravenous iron has been summarized as ‘6H-syndrome’ (high FGF23, hyperphosphaturia, hypophosphataemia, hypovitaminosis D, hypocalcaemia and secondary hyperparathyroidism)39. Other acquired forms of FGF23-related hypophosphataemia should be considered, including alcohol-induced FGF23 syndrome and ectopic FGF23 syndrome, which can occur as a rare complication in patients with advanced malignancies, especially prostate cancer and lung cancer40,41,42.
A 2022 study that used the Immutopics ELISA to evaluate plasma FGF23 levels in a cohort of paediatric and adult patients with hereditary and acquired forms of renal hypophosphataemia, reported that a 27-pg/ml cut-off value of intact FGF23 was 100% sensitive and 100% specific for distinguishing FGF23-mediated hypophosphataemia from FGF23-independent hypophosphataemia36. Similarly, in Japanese patients, a 30-pg/ml cut-off value of intact FGF23 in plasma enabled discrimination between XLH and non-FGF23-related forms of renal hypophosphataemia using another ELISA (Kainos assay)2. The reference values for healthy children established by the Hannover Reference values for Pediatrics study (which used the Immutopics assay) suggest that a cut-off value for intact FGF23 of 27 pg/ml corresponds to z-scores (percentiles) of −1.19 (12th), −1.73 (4th) and −1.87 (3rd) in children aged 1, 5 and 15 years, respectively29. This finding supports the concept that even ‘low–normal’ intact FGF23 levels suggest a FGF23 excess in the setting of concomitant hypophosphataemia. However, measurement of FGF23 levels is not available in many centres and results are influenced by the type of test that is used.
The first diagnostic approach for a patient with rickets or osteomalacia should be to exclude metabolic acidosis, renal diseases that cause non-selective tubular wasting that is not restricted to phosphate, and calcipenic diseases that cause hypophosphataemia secondary to hyperparathyroidism (Fig. 3). Once these diseases have been excluded, urinary calcium excretion and 1,25(OH)2D levels help to distinguish between FGF23-mediated diseases and primary tubular phosphate wasting due to pathogenic variants in the sodium-dependent phosphate transporters NPT2a and NPT2c, which are characterized by increased levels of 1,25(OH)2D and hypercalciuria secondary to suppression of FGF23 by hypophosphataemia and suppressed PTH serum levels43. Importantly, FGF23 levels are not informative in patients treated with oral phosphate or active vitamin D. A family history of X-linked inheritance with full penetrance in female carriers strongly supports the diagnosis of XLH. If a genetic diagnosis of XLH has been made in an index patient, confirmatory genetic testing may not be necessary in other family members with overt phenotypes.
Since the publication of our 2019 guideline, the diagnostic value of cone beam computed tomography to assess dental status and detect and analyse peri-apical and alveolar bone defects in patients aged >6 years has been established44,45 (Box 2).
Evaluation of responses to therapy
To guide the management of children and adults with XLH, we developed definitions to evaluate responses to therapy (Box. 3).
Before the introduction of burosumab, children with XLH were treated with oral phosphate and active vitamin D as standard. Active vitamin D (calcitriol or alfacalcidol) is given in addition to oral phosphate to counteract calcitriol deficiency, prevent hyperparathyroidism and increase phosphate absorption in the intestine. This treatment should be individualized and titrated based on clinical and radiological signs of rickets, serum alkaline phosphatase (ALP) and parathyroid hormone (PTH) levels and urinary calcium excretion17. Early treatment results in superior outcomes46,47,48,49,50. Studies have shown that in a proportion of children, oral medication improves bone pain, radiological signs of rickets and serum ALP levels within 12 months6,51,52. These treatments promote growth with rates within the normal range (>25th percentile for sex and age) and can gradually improve leg deformities as well as dental health47,48,49,53,54,55,56. Nonetheless, in most patients, complete response is unlikely and radiological lesions do not regress completely, with ALP often remaining above the upper limit of normal for age. Oral phosphate supplements and active vitamin D can also promote hyperparathyroidism and cause nephrocalcinosis in a dose-dependent manner, which limits dose escalation and thus efficacy17,57,58. Unlike therapy with burosumab, fasting phosphate levels are not restored by oral supplements and should not be used to target therapy5,6.
The indication for treatment of adults with XLH with oral phosphate and active vitamin D is more controversial than for children. Although some asymptomatic adults are advised to discontinue therapy after completing growth, several studies have shown improved long-term outcome in asymptomatic patients who have continued treatment into adulthood. However, this improvement may reflect confounding as patients who are more adherent to therapies in general may have better outcomes. Treatment can be considered for patients with substantial bone and muscle pain, pseudofractures, before and after orthopaedic surgery, oral manifestations (abscesses, periodontitis), evidence of osteomalacia (for example, increased total or bone-specific ALP levels) and during pregnancy. Treatment with active vitamin D and phosphate supplements has been shown to improve musculoskeletal pain and stiffness, to ameliorate osteomalacia and to decrease the frequency of dental infections and periodontitis7,46,59,60,61,62,63,64,65,66. No positive effects on hearing loss, osteoarthritis or enthesopathies have been observed5,6,65,66. In most studies, clinical improvement has been documented within 12–24 months after starting therapy. However, the potential benefits of phosphate and vitamin D therapy have to be balanced with the risk of severe hyperparathyroidism that can require surgery, as well as the risk of nephrocalcinosis.
In contrast to oral supplements, burosumab is a targeted therapy that directly inhibits the activity of FGF23 and thereby reduces renal phosphate wasting and increases endogenous 1,25(OH)2D levels. Increased tubular maximum reabsorption of phosphate per glomerular filtration rate (TmP/GFR) and serum phosphate concentrations have been observed within 1 week after initiation in children and adults, and can be used to adapt dosages18,19,20,24. Likewise, in the majority of patients of all ages, serum ALP levels improved within 6 months and normalized within 12 months of burosumab treatment and musculoskeletal pain was rapidly reduced.
In growing children, improvement in the severity of radiological rickets was observed after 1 year of burosumab therapy. However, catch-up growth was low (approximately +0.1 s.d per year) and an improvement in bone deformities was generally only observed after 2–3 years18,67,68. In adults, significant improvement in stiffness was observed after 24 weeks of treatment and confirmed at 48 weeks, whereas improvements in physical function and pain did not achieve statistical significance after Hochberg multiplicity adjustment24,69. Healing of osteomalacia (assessed using serum bone-specific ALP and bone biopsies) and pseudofractures have also been documented after 48 weeks of burosumab therapy69,70. Evidence for the efficacy and safety of treatment with oral phosphate in combination with active vitamin D and burosumab in patients with XLH is further detailed below.
Follow-up of patients with X-linked hypophosphataemia
Our guidelines for the follow-up of patients with XLH include new recommendations for the monitoring of those who are receiving burosumab treatment (Table 1 and Box 4).
Although treatment with oral phosphate supplements and active vitamin D improves symptoms in most patients with XLH, a complete response is generally not achieved6,46,47,48,49,50,51,52,53,54,55,56. Dose adjustments of oral supplements are based on improvement of ALP levels and of clinical and radiological signs of rickets or osteomalacia. Serum phosphate levels remain low despite oral supplementation and are not a target for adjusting therapy, which requires the primary goal of achieving a satisfactory clinical response to be balanced with the risk of developing nephrocalcinosis and hyperparathyroidism5,6,17.
Burosumab directly inhibits FGF23 activity and therefore improves urinary phosphate reabsorption and 1,25(OH)2D levels18,19,20,24. Phosphate and TmP/GFR levels increase rapidly after starting burosumab and can be used to inform dose adjustment during the first few months of treatment until other signs of clinical response can be assessed18,19,20,24.
In patients without previous long-standing treatment with active vitamin D and phosphate supplements, the risk of developing hypercalciuria, nephrocalcinosis and hyperparathyroidism when treated with burosumab is probably very low. Monitoring these parameters is therefore less important in patients treated with burosumab than in those receiving phosphate supplements and active vitamin D. However, we suggest monitoring of 1,25(OH)2D levels at least yearly as they may increase above the normal range, which may promote hypercalciuria71. Measuring FGF23 concentrations to monitor treatment efficacy is not recommended because burosumab interferes with the analytical assays72,73.
Treatment of children with X-linked hypophosphataemia
Our recommendations for the primary treatment of XLH in children have changed fundamentally compared with our 2019 guideline owing to data from an RCT that compared burosumab with phosphate supplements and active vitamin D67 (Box 5). In addition, the results of real-world studies and long-term data from the initial clinical trials of burosumab treatment in children with XLH are now available68,71,74,75.
Skeletal manifestations
An RCT that included 61 patients aged 1–12 years with XLH and persistent rickets (defined as a rickets severity score (RSS) of ≥2.0), compared 40–64 weeks of burosumab treatment with treatment with oral phosphate and active vitamin D67. The burosumab group showed higher rickets healing rates and greater improvements of radiographic global impression of change (RGI-C) total score, RSS, lower-limb deformity score, ALP levels, serum phosphate, TmP/GFR, height and 6MWT than the oral phosphate and active vitamin D group. A post hoc analysis showed that burosumab was superior to oral phosphate and active vitamin D with respect to RGI-C for rickets score and improvement of ALP serum levels at 40 and 64 weeks, irrespective of high or low phosphate and vitamin D doses74.
As children with XLH and mild–moderate rickets (RSS <2.0) and adolescent patients with XLH were excluded from the RCT, whether burosumab treatment is also superior to oral phosphate and active vitamin D in these groups is unclear. An observational study that included 65 children and 28 adolescents (>12 years) with XLH, most of whom had had previous long-term treatment with active vitamin D and phosphate supplements, reported that 12 months of burosumab treatment normalized ALP serum levels in about 80% of both cohorts, suggesting a substantial improvement of rickets in these patients. Adolescents seem to require lower doses of burosumab per body weight than children to obtain the same clinical response.
In 2018, burosumab was approved by the FDA and by Health Canada for the treatment of children with XLH from the age of 6 months. In 13 patients aged 1–4 years with XLH and an RSS of ≥1.5, a period of 64 weeks of burosumab treatment improved serum phosphate levels, RSS and RGI-C total score76. Efficacy and safety data for burosumab in infants with XLH are not yet available. However, a trial in participants aged <12 months has been completed and the results are expected in 2025.
Growth
Linear growth in children with XLH treated with oral phosphate and active vitamin D is reportedly highly variable, but typically well below average. In a large multicentre study of 228 children who were treated with oral phosphate and active vitamin D, the median height z-score progressively declined from around 0 at 3 months of age to around −1.6 at 2 years of age77. Single-centre and national studies have reported similar progressive declines in growth, with median z-scores between −1 and −2 at the end of the respective follow-up periods6,48,78.
In the RCT in children with XLH and persistent rickets, a small but significant improvement in mean height z-score at week 64 was reported in the burosumab group compared with the oral phosphate and active vitamin D group (0.17 (standard error 0.07) versus 0.02 (standard error 0.04), P = 0.049)67. However, a single-centre retrospective study of 55 children with XLH who were treated with burosumab for a median of 3.3 years reported no significant difference in the median height z-score at last follow-up (−1.07) compared with the beginning of treatment (−1.19)75. Although this study failed to show catch-up growth with burosumab, the participants did not experience a progressive decline in height z-scores over the duration of treatment. Stable height z-scores after 1 year of burosumab treatment were also reported in another retrospective study79. These findings indicate that early burosumab therapy prevents the progressive decline in height z-scores that is often observed with treatment with oral phosphate and active vitamin D. Under burosumab therapy, the height z-scores appear to be stable or to improve slightly.
Oral manifestations
The RCT in children with XLH and persistent rickets did not report a beneficial effect of burosumab treatment compared with oral phosphate and active vitamin D on the occurrence of dental abscesses67. Considering the time required for tooth formation, the duration of treatment (64 weeks) may have been too short to show a clinical benefit. Moreover, the dental infections that were reported in this RCT occurred in teeth that had mineralized before the onset of burosumab treatment. Primary teeth mainly form during pregnancy and the early months of life, whereas permanent teeth progressively form from birth onwards80. A post hoc analysis of the RCT reported that young children (aged <5 years) who were treated with burosumab were less susceptible to dental abscesses than those who were treated with oral phosphate and active vitamin D (abscesses in 0% versus 25%, respectively)74. By contrast, among older children (age 5–12 years), dental abscesses were reported in 53% of those treated with burosumab but in none of those treated with oral phosphate and active vitamin D. These findings suggest a window of opportunity during tooth formation for burosumab treatment to limit the risk of dental abscesses.
A prospective case–control study in 10 children with XLH aged 8.8 ± 3.8 years at initiation of burosumab therapy showed persistence of enlarged pulp chambers, a hallmark feature of XLH, in the permanent mandibular molar after 3 years of this treatment81. However, only one patient had recurrent abscesses involving a deciduous molar tooth during the 3-year follow-up period. A monocentric retrospective study that included 71 children with XLH (38 on oral phosphate and active vitamin D (mean age 2.92 ± 3.0 years at XLH treatment onset) and 33 on burosumab therapy (mean age 8.36 ± 3.81 years at XLH treatment onset)) showed that burosumab therapy for an average of 3.2 years was associated with a significant decrease in the number of dental abscesses compared with oral phosphate and active vitamin D; burosumab treatment was associated with 0.01 ± 0.03 abscesses per month of follow-up whereas treatment with oral phosphate and active vitamin D was associated with 0.04 ± 0.05 abscesses per month of follow-up (P = 0.04)82. None of these studies reported an impact of treatment on the occurrence of dental caries or maxillofacial cellulitis. Thus, in young children, burosumab might have a small beneficial effect for the prevention of dental abscesses compared with oral phosphate and active vitamin D. More prospective data are needed to fully assess the potential beneficial effects of burosumab on dental health.
Quality of life
Despite long-term use of oral phosphate and active vitamin D, children with XLH experience a substantial disease burden, including pain and impaired physical functioning that persists throughout childhood59,83,84. A study that assessed health-related quality of life in children with XLH receiving oral phosphate and active vitamin D, reported anxiety and depression in ~25% and problems with walking and self-care in ~50–60%85. In the RCT in children with XLH and persistent rickets, treatment with burosumab enhanced health-related quality of life by reducing bone pain and fatigue scores and improving physical health scores at 40 and 64 weeks versus baseline, whereas no improvement was noted in patients who continued treatment with oral phosphate and active vitamin D86.
Safety
In the RCT in children with XLH and persistent rickets, treatment-emergent adverse events (TEAEs) that were considered to be related to the treatment by the investigator were more frequent in the burosumab group (59%) than in the oral phosphate and active vitamin D group (22%)67. These TEAEs were mostly injection site reactions, were mild in severity and resolved within a few days. No between-group difference in serious TEAEs was reported (9% versus 10%), and no TEAE led to study or treatment discontinuation or death. The incidence of drug hypersensitivity was higher in the burosumab group (38%) than in the oral phosphate and active vitamin D group (19%). This safety profile was consistent with those of phase II studies of 64 (ref. 18) and 160 weeks68 of treatment, with 56–73% of TEAEs related to burosumab. Similarly favourable safety profiles were observed in single-group studies of burosumab treatment76,87. No emerging or worsening cases of nephrocalcinosis, hypervitaminosis D or hyperparathyroidism have been noted in any burosumab treatment study. By contrast, therapy with oral phosphate and active vitamin D is well known to cause secondary and tertiary hyperparathyroidism and nephrocalcinosis, as well as frequent gastrointestinal adverse effects5,6,88,89.
Management of burosumab
In the clinical trials of burosumab therapy in children with XLH, the primary aim was to achieve substantial healing of rickets (assessed using the RSS) within 40 weeks of treatment by raising phosphate serum concentrations to the lower normal range within the first few months. In clinical practice, questions arose as to what extent serum phosphate levels need to be normalized in children with XLH to achieve substantial healing of rickets, and whether regular X-ray examinations are necessary to monitor the success of burosumab therapy. Data from real-world studies are now available that provide important information for adjusting the burosumab dosage and monitoring of treatment response in children and adolescents with XLH71,75 (Box 6). We also provide advice on burosumab dosing during the transition to adult care.
Initial treatment and tailoring
Pharmacokinetic studies in paediatric and adult patients with XLH showed a direct dose–response relationship of burosumab over a dose range of 0.1–2.0 mg/kg body weight90. The half-life of burosumab in children and adults is approximately 19 days and the highest serum concentrations of burosumab and parallel increases in serum phosphate and TmP/GFR levels are achieved 7–11 days after injection91. When evaluating serum phosphate levels, the interval of blood sampling from the last burosumab injection should always be considered; we suggest an interval of 7–14 days. The dose of burosumab should not be increased more frequently than every 4 weeks, as steady state can only be assumed after 8 weeks of stable dosing91.
In clinical practice, children and adolescents with XLH demonstrate high variability in the dose required to improve serum phosphate and ALP levels71,75. In clinical trials in which the burosumab dose was tailored to raise serum phosphate into the lower normal range (>1.032 mmol/l (3.2 mg/dl), the mean dose after 40 weeks was 0.8 mg/kg, which resulted in normal phosphate levels in about two-thirds of patients and substantial radiological improvement of rickets in the vast majority18,67,76.
In the German XLH registry, the median burosumab dose at 12 months ranged from 0.2 to 2.0 mg/kg and was significantly lower in adolescents than in children (0.7 vs 1.1 mg/kg body weight, P < 0.01). Despite persisting mild hypophosphataemia (serum phosphate z-score less than −2.0) in about half of patients, serum ALP was normalized in 80% of patients71.
In a retrospective cohort study in the UK, about half of patients did not achieve normal age-related plasma phosphate concentrations after a median burosumab treatment of 3.3 years75. However, ALP levels at the last observation did not differ between groups with normal or below-normal phosphate levels and were normalized in the vast majority of patients. Moreover, patients with subnormal plasma phosphate levels had a higher tubular reabsorption of phosphate than those with normal plasma phosphate, arguing against ongoing urinary phosphate wasting. These findings suggest that complete normalization of serum phosphate is not necessary to achieve a substantial improvement of rickets in children with XLH and might not always be feasible. We therefore suggest using serum phosphate levels to tailor burosumab dose during the first months of treatment and thereafter to use clinical and biochemical indicators of rickets activity, including bone pain, improvement of leg deformities in the growing child, ALP values in the age-related normal range and appropriate growth. If the response is not clear, a radiological evaluation should be considered.
In most countries, the proposed starting dose of burosumab is 0.8 mg/kg every 2 weeks. However, emerging evidence suggests that a dose of 0.4 mg/kg every 2 weeks may also be appropriate, as proposed in some UK, French and European guidelines92,93,94. In the seminal phase II clinical trial of burosumab in children with XLH, the starting dose was 0.2 or 0.3 mg/kg every 2 weeks18. Several groups have reported wide ranges of median weight-based burosumab dose at 12 months (0.2–2.0 mg/kg), suggesting that lower doses are sufficient in some patients71,95. A retrospective study from the UK reported a median dose of burosumab of 0.93 mg/kg (IQR 0.7–1.27 mg/kg) in 27 children with normal serum phosphate levels at 2.5–3.8 years after initiation of burosumab treatment75.
Self-administration of burosumab in children with XLH after routine training was shown to be safe and effective, and should therefore be encouraged to promote patient independence96,97. Monitoring FGF23 plasma levels is not helpful in patients on burosumab treatment as the available antibodies cannot discriminate between free FGF23 and FGF23 bound to burosumab, resulting in falsely high FGF23 plasma levels73.
Transition to adult dosage
Bone mass accrual continues after completion of longitudinal growth until young adulthood (around the mid-20s). The age at which bone growth is complete differs between sites, ranging from 16–18 years for the spine and femoral neck, and up to 35 years for the skull98,99,100,101. Balanced bone remodelling is maintained after the cessation of growth until the last decades of life, when a progressive decline of bone mass occurs. Termination of burosumab treatment may result in progressive re-occurrence of osteomalacia and, in our opinion, is likely to have a negative impact on the quality of life and bone mass accrual of adequately mineralized bone in patients aged 18–35 years. Attainment of a proper mineralized bone and peak bone mass is essential to reduce the risk of osteoporotic fractures in adulthood102. We therefore suggest continuing burosumab treatment when the patient reaches adulthood, at least until the mid-20s, the time of peak bone mass. If burosumab is not available, we suggest treatment with oral phosphate and active vitamin D.
Management of oral phosphate and active vitamin D
As no new evidence is available, our recommendations for the management of oral phosphate and active vitamin D in children with XLH remain the same as in our 2019 guidelines17 (Box 7).
Treatment of adults with X-linked hypophosphataemia
Data are now available on the long-term efficacy and safety of burosumab in adults with XLH, including data on bone histomorphometric measures of osteomalacia, phosphate homeostasis, pain, physical function and stiffness70,103,104,105,106. These data allowed us to make more specific recommendations for the indications and tailoring of burosumab treatment in symptomatic adults with XLH compared with our 2019 guideline17 (Box 8).
No additional evidence on the efficacy and safety of oral phosphate and active vitamin D in adult XLH has been published since the previous version of the guideline17. In brief, treatment with active vitamin D and phosphate supplements improves pain, osteomalacia and oral health (reduces the frequencies of periodontitis and dental abscesses) but does not prevent or improve hearing loss, osteoarthritis or enthesopathies. Weak evidence suggests that these treatments improve healing of post-traumatic or surgical fractures. Limited evidence suggests that treatment in asymptomatic adults improves outcomes. Taking daily active vitamin D and at least twice-daily oral phosphate supplements is burdensome for many adults and is associated with potential adverse effects6,7,46,49,59,60,61,62,63,64,65,66.
To date, no studies have directly compared the efficacy of burosumab with that of oral phosphate supplements and active vitamin D for the treatment of XLH in adults. However, in the pivotal phase III RCT of burosumab in adults with symptomatic XLH, most participants had a history of treatment with oral phosphate and active vitamin D and had persisting pseudofractures that healed when switched to burosumab24. In patients who were switched from oral phosphate and active vitamin D to placebo, pain, physical function and stiffness remained stable over 24 weeks with no substantial deterioration, suggesting minimal or no benefit from the previous therapy. Current evidence suggests that burosumab rapidly ameliorates bone pain and stiffness and improves fracture healing to an extent that is not usually observed with oral supplements. In the absence of studies that directly compare the two approaches, evidence for their efficacy can only be reviewed sequentially and compared indirectly. As musculoskeletal pain is often vague and the aetiology is difficult to determine, clinical assessment is recommended to exclude causes of pain that are not expected to respond to treatment, such as osteoarthritis, enthesopathy, degenerative disc disease and muscle strain or wear.
Skeletal manifestations
Burosumab has been shown to improve bone histomorphometric measures of osteomalacia70 and pseudofracture healing in adults with XLH69,70. In an open-label, single-group phase III trial that investigated the effect of burosumab 1 mg/kg every 4 weeks on measures of osteomalacia, 11 patients underwent paired iliac crest biopsies at baseline and 48 weeks of treatment. The results showed significant improvement in all osteomalacia-related histomorphometric measures at week 48 (mean percentage changes in osteoid volume/bone volume of −54%, osteoid thickness of −32% and osteoid surface/bone surface of −26%). The median mineralization lag time decreased by 83%. In parallel, markers of bone formation (procollagen type 1 N propeptide and beta-C-terminal telopeptide) were significantly increased at week 48 (ref. 70).
In the pivotal phase III RCT, 68 patients received burosumab 1 mg/kg and 66 patients received placebo every 4 weeks for the first 24 weeks, and then all patients received open-label burosumab for an additional 24 weeks24. At baseline, 32 patients in the burosumab group (47.1%) had 65 fractures (14 unhealed fractures and 51 pseudofractures), whereas 38 patients in the placebo group (57.6%) had 91 fractures (13 unhealed fractures and 78 pseudofractures). At week 24, 43.1% and 7.7% of fractures that were active at baseline were fully healed in the burosumab and placebo groups, respectively. By week 48, the healing rate of baseline fractures or pseudofractures in the burosumab group was 63.1% and the healing rate of these fractures in the placebo group that was switched to burosumab at 24 weeks was 35.2%69.
Some studies suggest that improvements in skeletal manifestations that are attained after 1 year of burosumab treatment can be maintained with continued treatment (for >3 years)103,107. However, a post hoc analysis that compared patients who discontinued burosumab after 96 weeks of treatment (n = 7) with those who continued treatment (n = 23) suggested that the beneficial effects on laboratory parameters, patient-reported outcome and functional assessment were lost when the treatment was stopped, but seemed to recover once the treatment was resumed103. Accordingly, the treatment effect of burosumab seems to be limited to the time of continuous treatment and the available data do not support sustained efficacy after stopping treatment. Continued treatment with burosumab therefore seems to be necessary for sustained clinical benefit.
Functional disability and pain
The pivotal phase III RCT reported that 24 weeks of burosumab treatment resulted in a significant decrease in the Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) stiffness subscale (difference −8.1 (SE 3.24), P = 0.012) compared with placebo24. However, no significant improvement in WOMAC physical function subscale was found with burosumab versus placebo after Hochberg adjustment (difference −4.9 (SE 2.48). There was no significant difference in brief pain inventory worst pain score and 6-min walk test (6MWT) between the groups either. Following the 24-week RCT, all participants received open-label burosumab treatment for an additional 24 weeks. At week 48, both groups showed sustained improvements from baseline in scores for patient-reported outcomes of stiffness, pain and physical function (WOMAC) and 6MWT (each P < 0.001)69. A further analysis that included extended follow-up for an additional 48 weeks reported significant improvements in the WOMAC, the brief pain inventory short form and the Brief Fatigue Inventory scores, and 6MWT in both groups at 48 and 96 weeks104. A post hoc analysis of the trial data also reported beneficial effects of burosumab on pain, stiffness and physical function at weeks 24, 48 and 96 in various clinically relevant subgroups105. In summary, the RCT showed decreased stiffness in response to 24 weeks of burosumab treatment compared with placebo and long-term observational data support improved physical functioning compared with baseline if treatment is provided for a longer time period.
Enthesopathy
In the pivotal phase III RCT of burosumab in adults, enthesopathy burden was defined as an exploratory end point. At baseline, 99.3% of participants had X-ray-confirmed enthesopathy; the mean calcaneal enthesopathy burden (sum of superior and inferior calcaneal spurs) was 5.64 cm (s.d. 3.12 cm) in the burosumab group and 5.54 cm (s.d. 3.1 cm) in the placebo group24. At week 24, total calcaneal enthesopathy burden was 5.90 cm (s.d. 3.56 cm) in the burosumab group and 4.07 (s.d. 2.38 cm) in the placebo group. Thus, the trial showed no evidence of an effect of burosumab on enthesopathy burden compared with placebo. This failure might be at least partly related to the short follow-up period. No studies have compared burosumab with oral phosphate and active vitamin D with regard to enthesopathies. Furthermore, no studies have compared the effects of XLH therapies on spinal stenosis or skull-base abnormalities.
Osteoarthritis
In the pivotal phase III trial of burosumab in adults with XLH, WOMAC was a key secondary end point. Burosumab significantly reduced the WOMAC stiffness subscale at 24 weeks compared with placebo. However, numerical improvements in the WOMAC physical function subscale were not statistically significant. No data are available on the impact of treatment with burosumab or oral phosphate and active vitamin D on osteoarthritis structural damage on X-rays or the course of osteoarthritis. Currently, there is no evidence that treatment with burosumab or oral phosphate and active vitamin D prevents or improves osteoarthritis in patients with XLH.
Oral manifestations
No prospective studies have compared the effects of burosumab versus oral phosphate and active vitamin D on oral manifestations of XLH in adults. The pivotal phase III RCT in adults with XLH did not report a beneficial effect of burosumab on the occurrence of dental abscesses compared with placebo24. During the initial 24-week double-blind treatment period, 9 of 68 patients (13.2%) in the burosumab group and 5 of 66 patients (7.6%) in the placebo group developed an abscess. In all patients, tooth formation was complete at the onset of the trial and the dental infections that were reported occurred in teeth that had mineralized before the onset of burosumab treatment. Considering the very limited remodelling of dental tissues that occurs in adults, the duration of burosumab treatment in this trial (24 weeks) may have been too short to show clinical benefits.
A retrospective study documented the oral health of adults with XLH who started on phosphate supplements and active vitamin D during childhood; 24 of these patients remained on this treatment and 4 switched to burosumab treatment as adults106. Those who started on phosphate and active vitamin D treatment before the age of 5 years had significantly better tooth health than those who started in later childhood or adolescence. However, those who received phosphate supplements and active vitamin D at the time of the last observation and those who were treated with burosumab were not analysed separately.
Another retrospective single-centre study investigated oral health in a cohort of 44 adult patients with XLH108. Compared with periods of no treatment, treatment with oral phosphate and active vitamin D was associated with a 55.9% reduction and treatment with burosumab was associated with an 86.4% reduction in the incidence of infections (P < 0.0001). A significant association between burosumab treatment and decreased incidence of infection was also found when treatment and non-treatment periods were compared for the same patients (P < 0.01). Consequently, the available data suggest that compared with no treatment, burosumab therapy can prevent or improve dental manifestations (infections, periodontitis, tooth loss and loss of dental implants) in adults with XLH. However, robust evidence for the efficacy of burosumab in improving oral health compared with phosphate supplements and active vitamin D is currently lacking.
Quality of life
Adult patients with XLH generally have a reduced quality of life, despite long-term treatment with oral phosphate and active vitamin D83,85. Data on changes in the quality of life after the initiation of burosumab treatment are lacking. However, data are available on WOMAC score and pain levels, which can be used as a surrogate for the quality of life. In the pivotal phase III RCT, treatment with burosumab for 24 weeks compared with no treatment led to significant improvements in patient-perceived stiffness, but improvements in other categories of the WOMAC score and Brief Pain Inventory worst pain score were not statistically significant24. However, patients who were in the placebo group and transitioned to burosumab at week 24 showed significant improvements in the WOMAC physical function subscale and the Brief Pain Inventory worst pain level at week 48. These studies suggest a benefit of burosumab treatment for quality of life in adults with XLH, although robust evidence is lacking24,69.
Safety
In the 24-week RCT, a similar safety profile (including injection site reactions and hypersensitivity events) of burosumab versus placebo was noted24,109. No treatment-related serious adverse events or meaningful changes from baseline in serum or urine calcium, PTH, nephrocalcinosis or anti-burosumab antibodies were reported24. In the 24-week, open-label burosumab treatment continuation period of the RCT, the rates of adverse events were similar in the burosumab group and the group that had previously received placebo, and no fatal adverse events or treatment-related serious adverse events were reported69. No significant change in nephrocalcinosis scores, serum PTH, calcium, eGFR or urinary calcium excretion occurred in either group. Two female participants discontinued the study owing to pregnancy and delivered healthy infants at term. In the 48-week open-label burosumab treatment continuation period after the 96-week phase III trial, no grade 4 or 5 TEAEs were noted103.
Management of oral phosphate and active vitamin D
No additional evidence on the optimal dosing of oral phosphate and active vitamin D in adults with XLH has been published since the 2019 guideline17. However, after extensive discussion, we reduced the recommended minimum daily doses of oral phosphate and active vitamin D and the maximum daily dose of oral phosphate because high phosphate doses are associated with adverse effects110, lower doses of oral phosphate and active vitamin D may be effective, and alternative treatment with burosumab has become available (Box 9).
Management of burosumab
As some patients may show an inadequate response to burosumab therapy (Box 3), we considered to what extent dosage adjustment could be beneficial (Box 10). The manufacturer suggests that the initial adult dose of burosumab should be 1 mg/kg every 4 weeks. This dose was derived from a phase I–II pharmacokinetic study in adult patients with XLH that used a four-step dose escalation up to 1.0 mg/kg every 4 weeks111. With a final dose ranging from 0.6 to 1 mg/kg every 4 weeks, approximately two-thirds of participants reached a serum peak level of phosphate >2.5 mg/dl (0.8 mmol/l), and one-third maintained through serum phosphate levels greater than the target of 2.5 mg/dl. None of the patients reached a phosphate level above the upper limit of the target (4.5 mg/dl (1.5 mmol/l)). In an extension study in 20 patients treated with 0.3, 0.6, or 1.0 mg/kg burosumab every 4 weeks, the dose was titrated to reach phosphate trough levels >2.5 mg/dl, resulting in a mean final burosumab dose of 0.8 ± 0.1 mg/kg at week 76, with one participant still receiving a 0.3-mg/kg dose. In the pivotal phase III trial, 68 patients received burosumab at a starting dose of 1 mg/kg with a planned 50% dose reduction if serum phosphate increased to >5.0 mg/dl (1.6 mmol/l), which occurred in 6 patients (9%) during the initial 24-week study period24.
Taken together, these data suggest that a dose of 0.5–1.0 mg/kg every 4 weeks is adequate to reach serum phosphate levels >2.5 mg/dl (0.8 mmol/l) in the majority of adults. With the suggested starting dose of 1 mg/kg, a minority of patients will require a dose reduction. No information is available on dose escalation for adult patients who do not achieve the target serum phosphate level using the recommended dose of burosumab. A case report described an adult patient who was non-responsive to a standard dose of 1 mg/kg every 4 weeks but showed clinical and biochemical improvement after the dose was increased to 90 mg every 2 weeks using the paediatric standard protocol for burosumab112. From an efficacy standpoint, most adult studies have shown sustained improvements in biological bone markers, pain, stiffness, bone healing and patient-reported outcome measures using a burosumab dose of 1 mg/kg every 4 weeks24,69,107.
Prevention and management of hyperparathyroidism
Hyperparathyroidism is a common and challenging complication in patients with XLH, particularly among those who are receiving long-term phosphate supplementation, and may persist after switching to burosumab treatment14. In patients with XLH, hyperparathyroidism may be due to PHEX deficiency and the stimulation of parathyroid cells by FGF23 excess, low 1,25(OH)2D levels, phosphate supplements, inadequate calcium intake and/or vitamin D deficiency15,16,113,114,115,116,117,118. Hyperparathyroidism may further aggravate renal phosphate wasting and promote rickets or osteomalacia in these patients. In two large cohorts of paediatric and adult patients with XLH on longstanding treatment with oral phosphate and active vitamin D, persistent hyperparathyroidism was reported in 62% and 25% and hypercalcaemic hyperparathyroidism in 17% and 10% of patients, respectively; these adverse effects were associated with lower serum phosphate levels13,14.
As for other patients at an increased risk of bone disease, it is important to ensure an adequate age-related nutritional calcium intake and to supplement with native vitamin D (cholecalciferol or ergocalciferol) with the aim of maintaining 25-OH vitamin D levels within the desired target range of healthy individuals119,120 (Box 11). This management is especially important during periods of rapid growth, pregnancy and lactation. The European Food Safety Authority recommends the following age-related daily dietary calcium intakes: 7–11 months, 280 mg; 1–3 years, 450 mg; 4–10 years, 800 mg; 11–17 years, 1,150 mg; 18–24 years, 1,000 mg; adults aged above 24 years, 950 mg121. For infants aged 0–6 months, a global evidence-based consensus statement recommends a calcium intake of 200 mg per day to prevent rickets119. Treatment with oral phosphate should always be combined with active vitamin D to prevent secondary hyperparathyroidism and doses should be adjusted by reducing phosphate or increasing active vitamin D to maintain PTH levels, serum calcium and urinary calcium excretion within the normal range. Conversely, in the case of suppressed PTH levels, oral phosphate should be increased or the dose of active vitamin D decreased. In adult patients with a mild phenotype of XLH and persistent normocalcaemic hyperparathyroidism, active vitamin D might be given without phosphate supplements if careful follow-up is provided.
Burosumab treatment resulted in sustained improvements in 1,25(OH)2D levels in paediatric and adult patients with XLH, and no significant changes in PTH levels were noted in those who were switched from oral phosphate and active vitamin D to burosumab18,24,67,68,69,70,71,74,103,107. However, patients with severe hyperparathyroidism were excluded from the XLH trials, and data on patients with XLH who were primarily treated with burosumab are lacking. In a real-world study, about one-third of paediatric patients with XLH showed persistently elevated PTH levels after switching from oral phosphate and active vitamin D to burosumab71. These elevated levels of PTH were not associated with inferior normalization of serum phosphate and ALP levels. Despite these limitations, burosumab appears to be better than oral phosphate and active vitamin D for the prevention of (progressive) hyperparathyroidism in patients with XLH.
Adjuvant treatment with the calcimimetic cinacalcet was shown to lower serum PTH and FGF23 levels, and thereby increase TmP/GFR and serum phosphate levels in patients with XLH receiving oral phosphate and active vitamin D116,122,123. Off-label use of cinacalcet may therefore be considered in patients with XLH showing persistently elevated PTH levels despite the above measures. However, data on the use of cinacalcet in combination with burosumab in patients with XLH are lacking. Cinacalcet is contraindicated in patients with hypocalcaemia and is associated with increased QT intervals, which require adequate assessment and monitoring124. Parathyroid resection should be considered as first-line therapy in patients with persistent hypercalcaemic hyperparathyroidism as this disorder is associated with nephrocalcinosis and reduced kidney function13,14. Hypercalcaemic hyperparathyroidism may persist or recur after parathyroid surgery in 10% of patients with XLH13,14.
Treatment with recombinant human growth hormone
Progressive disproportionate short stature is a hallmark of XLH in children and often persists despite treatment with oral phosphate and active vitamin D or even with burosumab48,68,75. Treatment with recombinant human growth hormone (rhGH) can be considered for these children (Box 12). The hypotheses that have been proposed to explain growth impairment in XLH are complex and include impaired availability of phosphate for terminal chondrocyte apoptosis, suppressive effects of FGF23 excess on growth plate chondrocytes, a primary effect of the mutated PHEX gene, which is expressed in proliferating and hypertrophic chondrocytes, or a combination of the above8,125,126. Chondrocyte columns are disorganized in Hyp mice, with over-activation of the ERK and MAPK signalling pathways. Inhibition of the MAPK pathway, as observed when FGF23 is antagonized, improves the growth plate architecture127.
In children with XLH and short stature who are receiving oral phosphate supplements and active vitamin D, treatment with rhGH transiently increases serum phosphate levels and produces a sustained increase in height z-scores for up to 3 years128,129,130,131,132,133. Prepubertal children usually respond better to rhGH than pubertal patients. However, a follow-up analysis of a 3-year RCT in short children with XLH showed no clear beneficial effect of rhGH treatment on final height compared with no rhGH treatment134. Conversely, a retrospective study of 34 short children with XLH receiving oral phosphate and active vitamin D reported that rhGH treatment for a mean of 4.4 ± 2.9 years resulted in a significant increase in final height z-score compared with the z-score for height before the start of rhGH treatment (−1.3 ± 0.9 vs −2.4 ± 0.9, P < 0.001)78. However, the final height z-score of these patients did not differ from that of 29 children with XLH who were not treated with rhGH owing to sufficient growth or who chose not to receive this therapy (−1.3 ± 0.9 vs −1.2 ± 1.1, P = 0.7).
Limited data are available on the effects of rhGH in children treated with burosumab. An observational study in children who were switched from oral phosphate and active vitamin D to burosumab reported that treatment with rhGH resulted in an increase in height z-score (0.2 ± 0.1) during the first year after the treatment switch, whereas no significant improvement in height was observed in participants who did not receive rhGH79. Taken together, the available data suggest that in short children with XLH, treatment with rhGH might improve final adult height, regardless of whether they are receiving burosumab or oral phosphate and active vitamin D.
Treatment of musculoskeletal symptoms
No new evidence on the treatment of musculoskeletal symptoms of XLH has been published since our 2019 guideline17. However, we have added a new recommendation to highlight the importance of involving a pain clinic to ensure multidisciplinary pain management as recommended for other causes of chronic musculoskeletal pain135,136 (Box 13).
Management of pregnant and lactating patients
The potential beneficial effects of treatment with oral phosphate and active vitamin D in pregnant and lactating patients were outlined in our previous guideline17. However, very limited data are available on the use of these therapies or burosumab during pregnancy. According to the product information, studies in animals have shown reproductive toxicity of burosumab. Use of burosumab is therefore not recommended during pregnancy or in sexually active patients of childbearing potential who are not using contraception (Box 10, Box 14). Whether burosumab is excreted in human milk in substantial amounts is unknown, so a risk of harmful effects in breast-fed newborns or infants of lactating patients on burosumab treatment cannot be excluded. However, low breast milk levels compared with serum levels of monoclonal antibodies were reported in patients treated with these therapies for other diseases and burosumab should be mostly degraded in the gastrointestinal tract of the infant137. Burosumab treatment may therefore be considered for breast-feeding patients with severe XLH-related symptoms who are expected to respond to burosumab.
Orthopaedic management
Leg deformities that result from impaired bone mineralization are a hallmark of XLH in children (Fig. 1). These deformities often persist or even progress during treatment with phosphate supplements and active vitamin D, necessitating corrective orthopaedic surgery in about one-third of patients48,138,139. The availability of burosumab has fundamentally changed the approach to preventing and managing leg deformities in patients with XLH.
Children with XLH
The approach to orthopaedic treatment in children on oral phosphate and active vitamin D has not changed since our previous guideline17 (Box 15). In comparison with treatment with oral phosphate and active vitamin D, treatment with burosumab may enable orthopaedic surgery to be delayed by several years, particularly in younger patients. Burosumab very gradually but steadily improves bone deformities by enabling growth of nearly normal bone tissue. Rickets continues to improve over time with burosumab treatment. In a study that included 52 children with open growth plates who received burosumab treatment, the RSS decreased by 0.9 ± 0.1 from baseline to week 160. The radiological global impression score (RGI-C) was 1.57 ± 0.1, 1.75 ± 0.1 and 1.89 ± 0.1 at weeks 64, 88 and 160, respectively. Similarly, the RGI-C lower-limb deformity score was 0.5 ± 0.1, 0.58 ± 0.1 and 1.05 ± 0.1 at the same time points68. These data indicate that rickets and leg deformities continue to improve for at least 3 years in children treated with burosumab. Orthopaedic surgery should therefore be delayed, if possible, to avoid unnecessary surgery or to enable less complex surgery.
Adults with XLH
Leg deformities in adults with XLH are the consequence of suboptimal treatment during childhood and adolescence, resulting in impaired bone mineralization. In contrast to children, adults with XLH are also prone to osteoarthritis and pseudofractures, which occur without trauma. Painful pseudofractures usually occur in the weight-bearing bones140. Our recommendations for orthopaedic management in adults are presented in Box 16.
A case series of patients with XLH reported that surgical limb correction before puberty was associated with a high risk of deformity recurrence139. A subsequent retrospective study of patients who underwent temporary hemiepiphysiodesis to correct coronal plane knee deformities reported a slower rate of deformity correction in patients with XLH than in a control group of patients without metabolic bone disease141. The patients with XLH also had a greater number of secondary procedures and a lower rate of achieving neutral mechanical alignment than those in the control group. The lower success rate may be partly due to insufficient metabolic control of XLH-associated rickets or osteomalacia. These findings suggest that surgical limb corrections should only be indicated in patients with optimized metabolic control (requiring interdisciplinary discussions) and severe distortion and mechanical complications or residual deformities after fusion of the growth plates.
Early-onset, clinically debilitating osteoarthritis involving the large joints of the lower extremities has been reported to affect more than 50% of patients with XLH by the age of 30 years and up to 85% with progressive ageing24,59,83,142,143,144. To what extent osteoarthritis is primarily a consequence of the effects of PHEX mutations and/or altered FGF23 signalling on cartilage development or a secondary complication of altered biomechanics following developmental rickets and persisting deformities remains unclear145. Total knee arthroplasty and total hip arthroplasty can be beneficial in patients with XLH and severe osteoarthritis, but joint replacement in this population can be technically difficult and should be performed by surgeons with experience in treating skeletal dysplasias and particularly XLH146. Cementless fixation of total hip arthroplasty has been reported to be feasible147.
Pseudofractures are not a consequence of acute trauma but an indication of longstanding suboptimal treatment resulting in osteomalacia in combination with abnormal mechanical stress in the case of limb deformities. Consequently, surgery is not warranted and data support high rates of healing once treatment of the metabolic condition is optimized24.
Management of oral health
The beneficial effects of treatment with oral phosphate and active vitamin D on oral health (reduced frequency of dental abscesses and periodontitis) in paediatric and adult patients with XLH were outlined in our previous guideline17. Close dental monitoring and other measures, including sealing of pits and fissures in children and supportive periodontal therapy in adults, are also important (Box 17).
A post hoc analysis of the RCT in children with XLH and persistent rickets67 reported that among participants aged <5 years, none of those in the burosumab group and 25% of those in the oral phosphate and active vitamin D group developed dental abscesses148. By contrast, among children aged 5–12 years, dental abscesses were reported in 53% of those treated with burosumab, whereas no abscesses were reported in those treated with oral phosphate and active vitamin D. A monocentric retrospective study reporting dental follow-up of 71 children with XLH showed that burosumab therapy for an average of 3.2 years was associated with a significant decrease in the number of dental abscesses compared with treatment with oral phosphate and active vitamin D82.
A retrospective analysis of oral health data from 44 adults with XLH reported that those who were treated with burosumab developed fewer endodontic infections than those who were not treated or who received phosphate supplements and active vitamin D108.
These initial reports suggest that in the paediatric population, especially in young children, burosumab might be superior to oral phosphate and active vitamin D for the prevention of dental abscesses. In adults, some evidence suggests a superior effect of burosumab versus oral phosphate and active vitamin D for prevention or amelioration of oral manifestations.
Management of hearing problems
As no new evidence on the management of hearing in patients with XLH is available, our recommendations remain the same as in our 2019 guideline17 (Box 18).
Management of neurosurgical complications
No new evidence on the management of neurosurgical complications in patients with XLH has been published since our previous version of the guideline17 (Box 19). In brief, craniosynostosis, Chiari malformations and syringomyelia have been described as complications of XLH149. Although craniosynostosis is fairly common in this population, surgery is rarely required. As for patients with XLH on oral phosphate and active vitamin D, those treated with burosumab who present with neurological clinical symptoms should undergo fundus examination and/or brain imaging17. To date, no data are available on the impact of burosumab therapy on neurosurgical complications.
Lifestyle recommendations
No new evidence on lifestyle for patients with XLH has become available since our 2019 guideline was published. However, we have added a new recommendation to avoid smoking and limit alcohol consumption in line with recommendations for the general population and for patients at an increased risk of chronic kidney disease to reduce long-term morbidity17,150 (Box 20).
Conclusions
XLH is a challenging disease that is associated with substantial morbidity and requires lifelong multidisciplinary care. Burosumab is the first-line treatment for symptomatic children with XLH; this therapy results in the normalization of phosphate homeostasis and healing of rickets in the vast majority of patients. Adults with XLH may be treated with oral phosphate and active vitamin D if musculoskeletal symptoms suggest the presence of osteomalacia. Burosumab treatment should be initiated in adults with XLH who have pseudofractures or insufficient musculoskeletal response to oral phosphate and active vitamin D or who experience substantial adverse effects of oral phosphate and active vitamin D. Important research questions remain to be addressed to enable optimization of guidelines for the treatment of this rare disease (Box 21).
Responses