Mineralization of alkaline waste for CCUS
Introduction
In recent centuries, atmospheric CO2 levels have risen from <250 ppm to roughly 420 ppm. Levels continue to rise, as we emit an additional 37 Gt of CO2 each year1,2. To avoid global temperature increases above 1.5 °C, carbon dioxide emissions need to be reduced to 18 Gt of CO2/year by 20301,2. Carbon capture, utilization, and sequestration (CCUS) has emerged as an important route toward achieving this goal.
Mineralization, or the reaction of alkaline materials with CO2 to form solid carbonate minerals, is a promising CCUS technology. The reaction is thermodynamically favorable and occurs spontaneously, albeit slowly. Current research efforts focus on enhancing the rate and efficiency of this reaction. Mineralization reactions can be classified as in-situ, involving the reaction of CO2 with subsurface geologic formations to form subsurface carbonates, or ex-situ, involving the reaction of CO2 with alkaline materials that have been extracted from the earth to form useful carbonate products.
In this review, we evaluate ex-situ technologies that utilize industrial waste as a feedstock for mineralization processes that produce solid carbonate products. This review is not comprehensive in terms of industrial waste feedstocks but rather focuses and highlights high-potential opportunities with significant potential to scale to high levels of CO2 sequestration. Each year, the industry generates enough alkaline waste materials to sequester 7.5 Gt of CO23. The products of these reactions can be used to make cement aggregate, supplementary cementitious materials, geopolymers, soil additives, and more, helping to offset CCUS costs. Figure 1 shows a large-scale pathway from alkaline waste material to carbonate products. Pioneering companies such as Arca, Rio Tinto/Talon Metals, and BHP have already begun pilot-scale mineralization work, but reaction optimization, improved measurement, reporting, and verification (MRV), and further analysis are needed before these technologies can be deployed on the gigaton scale3.
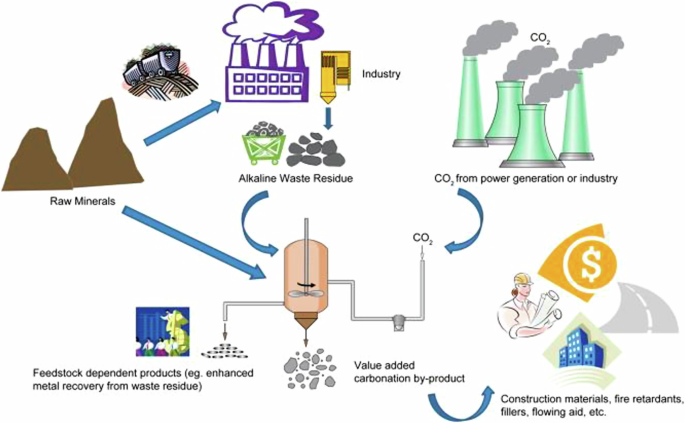
Reprinted with permission from Elsevier.
In this review, we have classified current technologies into the following categories: Direct gas/solid reactions involving diffusion of metal cations to react with gaseous carbon dioxide or dissolved carbonate in pore water in a single step, direct aqueous reactions which overcome kinetic barriers by dissolving the metal cations from a mineral where it reacts with dissolved carbonate, and indirect aqueous process which first leaches metal cations from a mineral in a separate step before adding a carbonate source in a second step. We present the state of the art for each category, evaluating both advantages and remaining hurdles associated with each approach.
Background
Some of the most promising waste sources for mineralization include ultramafic tailings. Ultramafic tailings have low silica content and a high Mg or Ca content and are often a byproduct of platinum group metal mining. Other highly alkaline tailings (i.e. from mining and ore processing industries targeting bauxite/red mud and Ni-laterite) are also of interest4,5,6. In addition to mine tailings, other industrial waste streams with alkaline chemistries that are receptive to CO2 mineralization include steel slag, cement kiln dust, fly ash, and other post-combustion materials from a variety of sources6,7,8. Sources like steel slag, cement waste, and ash often have multiple waste streams from different parts of the industrial process. Utilization of mineralized products will vary between these feedstocks, especially due to the roles of impurities and remaining secondary phases. One example is that large-scale carbonation of limestone waste can produce supplementary cementitious material that meets building and construction standards, which has the potential to greatly reduce emissions from cement manufacturing9. Several sources of compositional data are compiled in Table 1 to highlight the relative calcium, magnesium, silica, and impurity content. Table 1 also highlights the general composition of naturally occurring ultramafic rocks that make up a large portion of tailings.
The carbonate and bicarbonate products of this CO2 mineralization have a variety of industrial applications, offering additional value to support these mineralization processes. In many cases, the application of these mineral products could be a replacement for existing, carbon emission-intensive materials. For example, supplementary cementitious materials made from mineralized steel slag could reduce use of emission-intensive Ordinary Portland Cement. Utilization of mineralized products in place of emission-intensive products is estimated to have the potential to decrease CO2 emissions by 3.7 Gt/year4.
Waste stream contamination and toxicity must be addressed before the product can be utilized. While mineralization stabilizes heavy metal-containing waste against leaching (and is often utilized in tailings waste facilities for this purpose), materials containing toxic elements are not suitable for use as building materials or for other applications. Thus, any contamination must be addressed prior to use.
Direct mineralization processes include reacting solids with gaseous CO2 at elevated temperatures (direct gas–solid), as well as aqueous reactions with dissolved inorganic carbon (DIC) species generated by the interaction of water and CO2 (direct aqueous). In certain cases, phases in the waste stream can be completely converted to carbonates or bicarbonates by reaction with CO2, as shown in Eq. (1) for lime (CaO) present in cement kiln dust5:
However, in the majority of alkaline phases present in these waste streams there is some fraction of the stoichiometry that does not react with CO2, as is the case for the broad family of serpentine minerals often found in mine tailings with generalized stoichiometries (Mg,Fe,X)3Si2O5(OH)4, where X can be other diatomic elements and have the idealized CO2 mineralization reaction shown in Eq. (3)10. The more general reaction between magnesium silicates in water is shown in Eq. (2)11,12.
where even an incomplete reaction of the cations to form carbonate still results in exothermic reactions, with the ΔH of Eq. (2) equivalent to 64 kJ/molCO210.
However, in the case of phases that do not fully convert to carbonates, a reaction with CO2 can form a passivating layer that impedes the transport of the alkaline elements to a surface where they can react with CO2. Often, this passivation can be observed by two discrete kinetic regimes during direct gas–solid or direct aqueous mineralization13,14,15,16,17. Methods of alleviating this passivation effect in direct processes include reducing maximum passivation layer thickness by using small or porous particles, as well as elevated temperatures to moderately enhance transport kinetics13,18,19,20. Certain alkaline waste streams are conducive to pre-treatments or pre-waste processing that produce porosity, such as the firing of certain hydrate or hydroxide phases, which result in the evolution of H2O. Processes using firing pre-treatments are still considered direct and are especially common among ashes from wood pulp or related organic and/or coal-based feedstocks where the firing occurs during the production of the waste stream (in this case combustion of remaining carbon)15,16,21,22,23,24,25. Magnetic pretreatment to remove iron, a very common impurity, is effective and reduces the quantity of iron side products formed.
In contrast to direct processes, indirect mineralization processes rely on a discrete aqueous leaching step prior to mineralization, as seen in Fig. 2. Typically, this leaching step is performed at acidic conditions where the solubility of Ca2+, Mg2+, K+, and Na+ are high. Leaching from mineral phases requires a much lower pH than is feasible via carbonic acid produced by CO2 and instead relies on the addition of acids, including HCl, HNO3, acetic acid, sulfuric acid, citric acid, ascorbic acid, formic acid, and ethylenediaminetetraacetic acid (EDTA)26. When used on alkaline silicates or aluminates, this leaching step encounters a related passivation phenomenon to that found during direct carbonation reactions. Multiple methods have been reported for addressing this passivation, including the design of leachate solution as well as sonication26,27,28,29,30,31,32,33. The higher requirement of chemical additives in indirect processes can be a barrier for larger implementation, so much research exists around the regeneration of the required acids and bases or using a single, recyclable additive like ammonium salts.
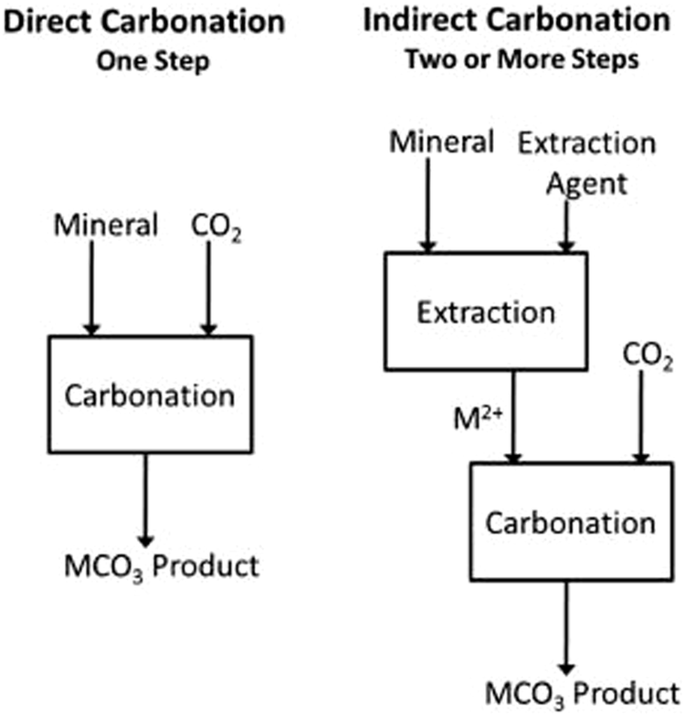
Reprinted with permission from Elsevier.
Indirect processes have a separate mineralization step after the leaching of soluble alkaline elements, which takes place at a higher pH where the solubility of the desired solid phases is much lower, typically 7–10 for bicarbonates and 10–12 for carbonates. These target pH regimes are closely related to solution concentrations of bicarbonate and carbonate ions. Targeting unique solubilities of carbonate products also allows for selective precipitation, which is quite important to understand to yield economically viable mineralization processes.
Water plays a critical role in CO2 mineralization processes. The reaction of CO2 and H2O results in a number of species including dissolved CO2 (aq), carbonic acid (H2CO3), bicarbonate ions (HCO3−), and carbonate ions (CO32−), with the relative concentrations of these species dependent on the pH of the solution as shown in Fig. 3. In aqueous processes, a high H2O vapor pressure is frequently used to increase the boiling point of water and enable more rapid reaction kinetics; however, increasing temperature decreases the solubility of CO2 and results in lower levels of DIC, including carbonate ions, CO32−, unless elevated pressures of CO2 are also used to compensate for thermal effects and maintain high DIC concentrations. H2O plays a critical role even in direct solid-gas reactions, with partial pressures of steam added to CO2 to greatly increase the kinetics of carbonate mineralization, most likely by modifying the surface chemistry of the solids and the surface reactions of CO26,22.
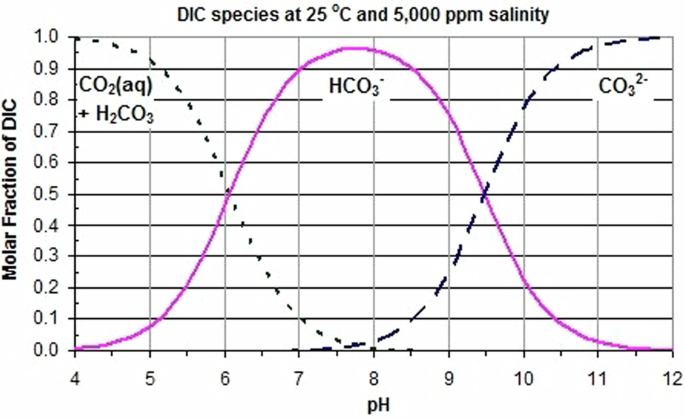
Reprinted from Creative Commons.
While many alkaline wastes are chemically similar, few demonstrations of CO2 mineralization have targeted more than one feedstock, leading to uncertainty in the transferability of results between feedstocks. In addition, the relative merits of applying direct gas–solid, direct aqueous, and indirect processes to different feedstocks depend on the degree of process intensification in terms of temperatures, pressures, and other variables required by that feedstock. Ultimately, there are processing pathways and feedstock-dependent tradeoffs between the percentage of CO2 mineralization that occurs relative to the idealized case and the intensity of the process parameters required. By examining instances of a range of processes and their application to different feedstocks, we endeavor to identify where parallels do and do not exist between the application of different mineralization techniques across different feedstocks.
Discussion
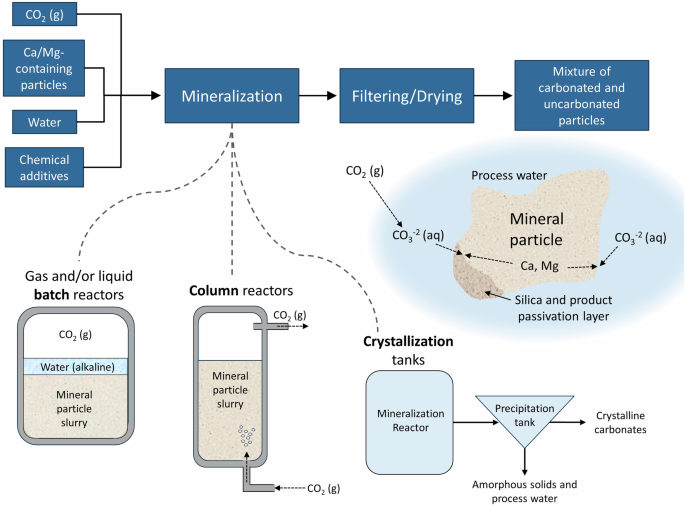
The two key advantages of direct gas–solid mineralization are high near-surface reaction rates and the potential for relatively low-energy intensity reaction conditions if the incomplete reaction of the alkaline elements is tolerable. Significant operating variables for direct gas–solid mineralization include temperature (from ambient to ≥600 °C), pressure, particle size, presence of moisture, presence of impurities, and reaction time. The basic reactions occurring at the mineral particle are shown in Fig. 4.
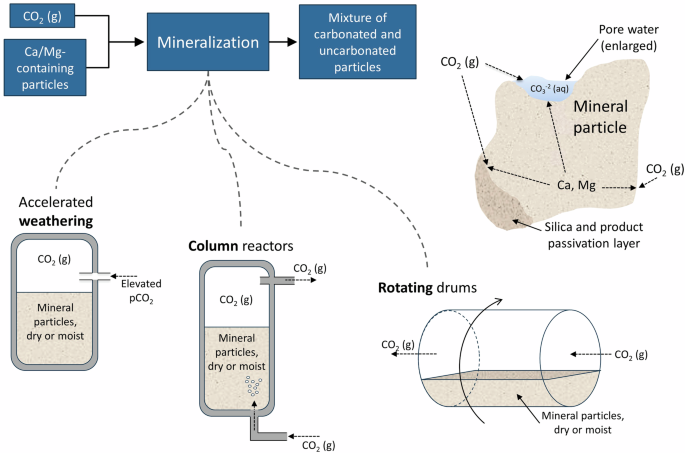
General flow diagram of direct gas–solid processes with main technologies and mineral particle carbonation chemistry shown.
Example applications of gas–solid mineralization to a range of alkaline feedstocks and a summary of relevant operating parameters are shown in Table 2. In experiments that tested a range of conditions, the conditions that yielded the best carbonation results are shown. The reaction mechanisms can generally be described as (1) the diffusion of CO2 into mineral pores, (2) the dissolution of alkaline ions from reactive phases in particles and diffusion to a surface containing un-reacted carbonate or CO2 species, and (3) the nucleation and growth of the carbonated phase34,35. In moist processes, additional carbonate species and metal cations are dissolved in the pore water, which increases available routes for mineralization. The carbonation reaction initially occurs quickly at the particle surface. This initial, near-surface, reaction stage can occur at roughly an order of magnitude faster than direct aqueous carbonation34,35. However, in minerals containing silicates or oxides, the carbonation reaction may result in the formation of layer(s) of un-reactive oxides or dense carbonate/bicarbonate product16,26,36,37,38,39 that limit diffusion of alkaline elements to reactive surfaces. Formation of these diffusion-limiting layers resulted in ≈3 orders of magnitude decreased rates of carbonate mineral formation, depending on passivation layer thickness and composition34,35. For example, in a 1 h and ≈50 °C reaction of fly ash, over 70% of total CO2 mineral formation occurred during the first two minutes34,35,40. Much of the research in this field focuses on improving reaction kinetics and uptake of CO2 by manipulating the above reaction parameters and pretreatment methods, of novel reactor design.
Multiple factors influence the kinetics and yield of direct gas/solid processes, including surface-to volume ratio of the solid particles, the temperature of the reaction, the CO2 partial pressure, the presence of water vapor, and the effects of sulfurous and ferrous phases. Due to the interplay between these variables, industrial alkaline waste with different form factors and stoichiometries may have different tradeoffs between process intensification (e.g. higher CO2 pressures and temperatures) and efficacy.
Due to the rapid near-surface reactions and the formation of passivation layers, the surface-to-volume ratio of feedstocks is a critical component of the kinetics of carbonation and, ultimately, the fraction of alkaline material that can be realistically carbonated on reasonable time scales6,7,20. Increasing the surface-to-volume ratio via small particle sizes or high porosity has both been shown to increase the carbonation mineralization kinetics. Particle sizes are as small as 48 μm have been tested for gas–solid carbonation and had over three times the fraction of alkaline elements carbonated as compared to the results of the same experiment with a particle size between 150–300 μm20. Some waste materials, like ashes, will naturally have comparatively smaller particle sizes, while decreasing the particle size of materials through an additional grinding pretreatment can have a high energy cost which should be considered in the sequestration capacity of the entire process26.
Elevated temperatures are frequently used to increase the kinetics and influence the phases formed during direct gas–solid mineralization19,39. However, the decomposition temperature of the target carbonated mineral serves as an upper limit for the process temperature. For example, chrysotile had the highest CO2 uptake at 375 °C, close to but not in excess of the decomposition temperature of MgCO3 in 1 bar of CO222. Elevated temperatures increase the rate at which alkaline elements diffuse across passivation layer(s), which helps mitigate its effect, but this benefit to kinetics is extremely dependent on the makeup of the passivation layer(s), grain structure, and the alkaline species.
Generally, increasing the partial pressure of CO2 increases the carbonation kinetics of industrial waste20,41,42. However, increased concentrations of CO2 only directly influence the kinetics of the near-surface reactions, with only weak interactions on the solid-state diffusion of alkaline elements governed by changing chemical potential across the passivation layer. Above the equilibrium CO2 pressure of the product phase, further increasing the pressure of CO2 has a greatly diminished impact on the mineralization kinetics once a passivating layer has formed43. The trend of increased mineralization kinetics at higher partial pressures of CO2 is maintained across humidities19,26,43,44. Similar to decreasing particle size and increasing temperature, the energy required to increase the CO2 partial pressure needs to be considered in the process energy balance because it can be quite significant.
The presence of moisture is important for accelerated gas–solid carbonation41,45. In mining waste and natural rock specifically, the calcium and magnesium are often bound in silicates that are not very reactive with dry CO246. The presence of water vapor in direct gas–solid reactions facilitates complex, multi-step surface reactions between the CO2 and solid phases, which can substantially increase the kinetics of carbonation reactions at the surface22. The optimal humidity is dependent on the temperature and specifics of the waste being mineralized. For coal fly ash, Brück et al. determined optimal moisture content in the gas stream to be around 20 wt%41,45,47. Tests on serpentine minerals (see Eq. (2)) in extreme humidities, such as 98%, observe an inverse relationship between increasing humidity and carbonate mineralization, likely due to decreased transport of CO2/displacement of CO2 away from the reactive surface48.
The presence of sulfur either in the feedstock or CO2 source (e.g. flue gas) can improve the carbon uptake of a process39,41. For example, gaseous SO2 up to 151 ppm improved the kinetics of mineralization of steel slag39. It is hypothesized that the sulfur undergoes a surface reaction with humidity to form sulfuric acid, which aids the leaching of alkaline minerals from silicates or other phases. Sulfates in mineral feedstocks, such as gypsum (CaSO4), readily convert into calcium carbonate in red mud37. Optimizing processes that involve sulfur will require balancing reaction kinetics and speciation with the impact of sulfur in emissions, in products, and on the durability of reactor materials49.
The presence of iron oxides has been shown to impede the kinetics of select gas–solid mineralization reactions. Thin layers of iron (III) hydroxide have been observed to form on the surface of particles, acting as a hydrophobic passivation layer16,19,41,43,46,50,51. Magnetic separation of iron before carbonation yields products with fewer impurities and possibly economically valuable magnetic by-products19.
Many different reactor designs have been studied to optimize gas–solid carbonation reactions for different variables and considerations, including accelerated weathering in piles, curing, fixed bed reactors, fluidized bed reactors, and rotating drum reactors42,48,52. The influence of the ratio of gas to solid material, which is unique to gas–solid reactions and not yet well understood, and batch vs continuous operation have been evaluated for different reactor types47,48.
Direct gas/solid mineralization has been demonstrated at varying degrees of process complexity and intensification, with accelerated weathering being the least complex and nearest ambient process. Accelerated weathering in piles mimics natural weathering but adds periodic watering, pile stirring, and increasing CO2 pressures to improve kinetics36,43. While the kinetics of carbonating mineral piles are poor compared to most reactor experiments, resulting in almost negligible carbonation below surface minerals, tailing pile mineralization has the potential to operate very cheaply and is possible under atmospheric conditions6,41,46,52. It is most commonly performed within the concrete industry because the resulting material is dense enough to meet mechanical standards for aggregate17,46,50.
Fixed and fluidized bed reactors flow a CO2 gas stream through the solid waste material for the duration of the carbonation experiment and offer a number of improvements over stationary gas–solid reactors20,40,42,46,48,51,53. Residence time, a process variable specific to flow reactors, plays a significant role in total CO2 uptake through mineralization46. First, the continuous flow of CO2 mitigates local CO2 depletion that can occur during batch processes as carbon is mineralized. The use of a fixed bed reactor to mineralize Ca(OH)2 did not show full conversion to CaCO3, which could be partially mitigated by using fluidized bed reactors because of the increased interactions between minerals and CO2 and mitigation of the passivation layer through particle-particle abrasion inherent in fluidized beds48. Lastly, fluidized beds have the potential to capture the heat released in the reaction, which can be captured or reused to decrease the total energy requirement of the process.
Several municipal solid waste incineration bottom ash (MSWI BA) mineralization demonstrations have used a continuous-feed rotating drum reactor47,54. The application of these rotating drum reactors to other feedstocks is poorly understood44,47,54,55,56. The impact of these reactors as compared to batch reactors is very similar to the improvements with fixed and fluidized beds as described above and increase the number of continuous flow reactors.
A diversity of pilot-scale gas–solid demonstrations have been performed41. Pilot-scale production of aggregate from cement kiln dust (CKD), wood ash, and paper ash carbonation was commercially viable, but further studies are required on their long-term resistance to environmental degradation, such as the impact of sulfur and chloride16,17. Additionally, pilot trials of gas–solid carbonation of coal fly ash were performed with a moisture-reducing drum, a heater/humidifier, and a fluidized bed reactor in series that could react with roughly 100–300 kg of fly ash. This trial sequestered a higher proportion of carbon dioxide in 1 h than the laboratory-scale experiment under very similar conditions, while successfully reducing the flue gas concentrations of mercury and sulfur40. However, this indicates sulfate and mercury carbonate phases may also exist in the products40. Large-scale gas–solid carbonation will still face challenges with material reactivity that will require balancing against the energy intensity of pre-treatments37. Gas–solid processes tend to be the simplest and lowest-cost methods of carbonating alkaline wastes. Material and capital costs approach zero for simple processes like accelerated weathering. An economic estimate of gas–solid carbonation from a 532 MW power plant with coal fly ash with a capacity of 207 kg CO2/t fly ash was about $11/t, which is slightly more expensive than estimates for geological sequestration ($7/t CO2), not factoring in the value of product carbonate phases40. There is less research on the separation and purification of products because gas–solid processes are often chosen for their relatively lower cost. Further research into this might improve the profitability of gas–solid processes, but the current focus revolves around forming products that do not require separation or purification, such as cementitious materials or aggregate20,41,48.
Direct aqueous carbonation processes are designed to overcome many of the kinetic limitations that gas–solid reactions face. The presence of bulk water allows for easier activation of mineral silicates through increased proton attack of the amorphous silica that allows for metal leaching, as seen in Fig. 557. The most significant kinetic limitations include carbon dioxide dissolution and conversion into carbonate ions, which is very pH sensitive, and diffusion of calcium and magnesium in a mineral to reactive sites. Many of the important operating variables are the same as for direct gas/solid mineralization but have differently sized impacts on an aqueous system. Table 3 gives an overview of many examples of direct aqueous processes.
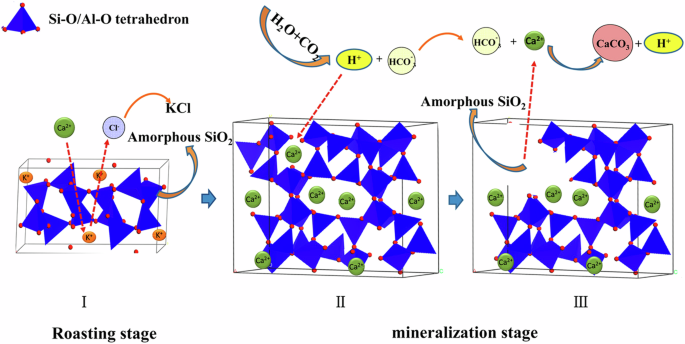
Reprinted with permission from Ye et al. 124. Copyright 2014 American Chemical Society.
The reaction mechanisms for aqueous carbonation of concrete waste, which can be generally applied to calcium-containing minerals, were determined to be (1) extraction of Ca2+ from a mineral, (2) CO2 dissolution into the aqueous phase, and (3) carbonation and precipitation of CaCO358,59. These mechanisms are represented in Fig. 6. Most experimental processes create a slurry that hydrates calcium oxide and silicates to produce Ca(OH)2 and calcium silicate hydrates. When CO2 is introduced, these intermediates are converted to CaCO3 and SiO232. Depending on the process design, either the calcium or the carbon dioxide dissolution and diffusion step can be rate limiting59,60. The mineralogy, morphology, and microstructure of each waste material are the most significant factors in determining carbonate mineralization capacity32. Pretreatment specific to aqueous mineralization includes tailoring a cooling process to promote crystallization of more reactive phases and microwave pretreatment, for example of serpentine into olivine32. While these were successful in increasing carbonation capacity in steel slag and red mud, respectively, pretreatment choices need to be made with respect to the energy balance of the process. One analysis determined microwave pretreatment was 4–8 times more energy intensive than thermal treatment21,61,62. An inert, porous passivation layer commonly forms on the surface of particles during carbonation, specifically a SiO2 rim, as calcium and magnesium are leached from the mineral63.
General flow diagram of direct aqueous processes with main technologies and mineral particle carbonation chemistry shown.
The influence of temperature is not as straightforward in aqueous carbonation as compared to gas solids but is limited to temperatures below the boiling point of water at operating pressure. Temperature affects kinetics, equilibrium values, CO2 dissolution, and calcium leaching as well as the phases and polymorphs formed31,34,64. High temperatures increase calcium and magnesium dissolution but decrease the solubility of CO26,7,31. The solubility of carbon dioxide is subject to Henry’s law, which describes a direct linear relationship between partial pressure and concentration of dissolved CO26. However, the constant in Henry’s law decreases with increasing temperature for carbon dioxide26. Steel slag, nickel mine tailings, gypsum, and coal fly ash have experimentally determined optimal carbonation to occur between 25 and 80 °C34,55,65,66,67,68,69. Higher temperatures can be successful if carbonate concentration is increased in another method, such as increasing CO2 pressure or introducing additional chemical carbonates21,70,71,72. For example, Huijgen et al. saw the highest carbon uptake at 175 °C but operated at 19 bar CO2. The addition of chemical carbonate species such as Na2CO3 is less applicable to the scope of this paper, but some research has indicated that NaHCO3 accelerates CO2 dissolution which could be interesting for further exploration to overcome dissolution limits6.
The liquid/solid ratio (L/S ratio) is very important in carbonate conversion because higher L/S ratios can inhibit mass transfer, decrease ionic strength, and reduce supersaturation and precipitation32,73. The decreased interactions between reactants at high L/S ratios cause a decrease in driving force that slows the reaction kinetics and decreases carbonation efficiency38,55. However, decreasing the L/S ratio increases the relative reactant mass which increases reaction time and CO2 removal38,59,68,72. Additionally, calcium and magnesium are more soluble in higher ionic strength solutions (low L/S ratios)71. Therefore, the L/S ratio should be selected to ensure enough liquid is present to dissolve sufficient carbonate and increase reaction kinetics without flooding the reactor. However, some studies considered CO2 dissolved in process water in their carbonation efficiency calculations and reported better results at higher L/S ratios. For mineralization studies, quantification of the carbon content in the product needs to be performed with X-ray diffraction (XRD) or thermogravimetric analysis (TGA)60,74. Alternatively, a few experiments first calculated the pressure drop due to CO2 dissolution in a representative system before using the overall pressure drop to calculate the CO2 pressure drop from mineralization67.
Increasing CO2 pressure increases the dissolved carbonate concentration according to Henry’s law and, in most cases, increases carbon mineralization6,74,75. It has been shown to increase both rate and overall carbon uptake31,76,77. Higher pressures (Table 3) have been observed to improve nucleation and growth kinetics, show influence on the precipitated calcium carbonate morphology, and sometimes inhibit crystal formation. Specifically, high pressure yielded larger crystal sizes and favored aragonite formation in gypsum samples76. The acidification from CO2 dissolution increases the solubility of both calcium and magnesium salts as well as calcium and magnesium carbonates, the latter decreasing precipitation29,31. Several studies have tried to mitigate this by adding a separate precipitation tank with a lower total pressure18,23,78,79. Using a gas stream with a lower CO2 content, as compared to a batch process at higher pressure, can still achieve good reaction kinetics if the L/S ratio is reduced such that diffusion rates and supersaturation increase55. Several studies have demonstrated good results with low pressure conditions59,66,76, and others have concluded that pressure had no impact on carbonation efficiency38,58,67. The gas flow rate, in those cases, controlled CO2 reaction kinetics. This implies that optimal CO2 concentrations exist in relation to both the calcium and magnesium content and the L/S ratio such that the rates of dissolution and carbonation are balanced55,59.
Surface-to-volume ratios are a key factor in direct aqueous mineralization processes. Small particle size, as small as 38 μm, favored mineralization for calcium species. An increase in particle size from 38 to 75 μm statistically significantly decreased uptake from 3.05 wt% to 1.42 wt%65. However, not all experimental studies with ore have observed this trend21,26,80, and so instead it is hypothesized that the increase in carbon uptake is due to the increase of specific surface area rather than particle size63. Pretreatment by microwaving or grinding increases specific surface area, although in practice this has also increased the exposure to air and, therefore, iron passivation21. Within one mineral sample, different categories of sizes of particles, separated by sieving, can have different chemical compositions which has led to experimental results revealing different trends than expected regarding particle size and sequestration74,80. For example, cement waste particles between 75 and 300 μm had the highest calcium dissolution rates, but it was determined that that size range of particles had a higher concentration of calcium80. Some studies conclude that decreasing particle size beyond a certain point has negligible effects, 75 μm as another review concluded26. Overall, small particle sizes on the order of μm to mm are important for carbonation rate and efficiency, but it is likely equally important what phases and impurities are present.
The role of sulfurous species in direct aqueous mineralization is complex and pH-dependent. In the case of direct aqueous carbonation of gypsum at alkaline conditions, sulfate products are common since the sulfur is fully oxidized and the alkalinity means highly soluble sulfuric acid will not form. Alternatively, sulfur dioxide in flue gas increases agglomeration of the precipitated calcium carbonate particles as it reacts with oil shale ash18.
Similarly, the presence of iron is also very limiting for carbonation efficiency because the iron oxide, hydroxide, and oxyhydroxide products contribute to passivation on particle surfaces and influence the morphology of final products60,81. In aqueous systems, more acidic pHs increase the production of ferrous oxide passivation81. While iron species can form carbonates, in oxidizing environments, they form Fe2O3 and Fe(OH)3 much more rapidly than FeCO3 and end up limiting water penetration into the particle. For this reason, some experiments magnetically remove iron during pretreatment, though this approach is limited by the magnetic properties of the initial ferrous phase(s)23.
A silica and carbonate passivation layer, normally a few nanometers thick, forms during aqueous carbonation of calcium and magnesium silicates, steel slags, red mud, and cement waste23,34,35,59,63,70,82. As metal cations leach to the particle’s surface, the SiO2 phases are left behind in a porous rim that impedes further metal diffusion71. This silica layer forms in tandem with the iron passivation layer discussed above, complicating potential strategies to mitigate passivation. Some studies have seen the formation of carbonate crystals on the surface of particles, covering the silica layer and un-reacted core, likely due to incomplete extraction or calcium saturation issues83. Complete removal of the passivation layer is possible with mechanical exfoliation, which refers to grinding or aggressive mixing24,70. However, it can be difficult or impossible to separate the surface layer and un-reacted core particles70. Several authors use the shrinking-core model to understand the formation of the silica layer. An important assumption in the shrinking-core model is that the speed of reaction is infinitely faster than the diffusion of reactants through the product passivation layer, so this model should not be applied to every scenario because the limiting reaction mechanism can change greatly depending on operating parameters34,59.
Chemical additives have been used to improve calcium and magnesium dissolution, increase CO2 solubility, decrease passivation layer formation, and control pH29,38,70,72,76,80,81. The type and concentration of chemical additives affect the precipitated carbonate morphology. Water is not a very effective leaching agent, so strategies to increase calcium and magnesium dissolution focus on increasing the ionic strength or manipulating the pH72,77,81. There are conflicting results on the use of NaCl for metal leaching, as some authors claim the chloride ions improve magnesium silicate solubility and others claim it has no effect21,70. A separate experiment hypothesized that proton and ligand dissolution mechanisms are additive which contributes to the improved dissolution of magnesium and calcium in higher ionic strength solutions23,29. It is agreed that NaCl can mitigate the production of the passivation layer because of its effect on silicate solubility70,81. The negatively charged surface of the silica attracts Na+, causing a steric effect that allows H2O more access to the Si–O bonds, which dissolves the silica passivation layer more rapidly70,81,84. A mineralization experiment with steel slag produced 0.98 wt% more MgCO3 using 0.7 M NaCl70. While most studies used 1 M NaCl, more research can be done on the optimal proton/ligand ratio to reduce chemical additive requirement as much as possible21,29. One study used ionic liquids (4 wt% inerts) to dramatically increase the leaching rate (52 times) but saw slower dissolution of CO281. Chemicals like EDTA, cyclohexanediaminetetraacetic acid (CDTA), and oxalate have been used to increase the dissolution of metals and solubility of CO238,72,76,77,81. Periodic watering with EDTA, CDTA, and oxalate improved CO2 uptake, magnesium dissolution, and mitigation of the iron passivation layer81. However, carbonic anhydrase in ultramafic mining residue reduced the sorption of CO2 into the water. NaCl and EDTA have had conflicting effects on carbonation efficiency in fly ash carbonation38,72. NaOH, triethylamine (TEA), and hexamethylenetetramine (HMT) have been specifically used to increase the alkalinity and improve carbonation efficiency because carbonates significantly decrease solubility at high pHs77. NaOH is a common alkalinity source because it has a shorter conversion time, yields higher purity products, is inexpensive, and yields smoother surfaces and crystallinity of CaCO3 as compared to ammonium salts76. It can also be recovered or regenerated76,77. Lastly, 3-(Cyclohexylamino)-1-propanesulfonic acid (CAPS) and 2-[2-Hydroxy-1,1-bis (hydroxymethyl)ethylamino]ethanesulfonic acid (TES) have been used as buffers because the carbonation reaction and precipitation are extremely pH sensitive and have negligible reactions with calcium80.
Generally, aqueous mineralization systems require a tank in which gas and liquid contact occurs. Both batch and flow reactors have been explored to understand the effects of different process design variables on carbonation efficiency. Some designs include a separate precipitation tank18,23,78. In the precipitation tank, the solubility of calcium and magnesium carbonates has been greatly reduced such that they precipitate in relatively higher proportions and purity23,78. For example, a carbonation reactor was operated at 3 MPa CO2, but the precipitation vessel was operated at atmospheric pressure CO2, which resulted in a 96% conversion of CaCO378. The solubility of carbonates can also be manipulated via pH change and different carbonates will precipitate at different pHs, allowing for separation of products. It is unclear if there is a trend in the effect that precipitation tanks have on energy balances, but they clearly form purer crystalline products79. Silica can polymerize in water to be extracted as well21.
Fixed and fluidized beds have been studied in direct aqueous systems34,85. Fluidized beds have better mass transfer as compared to fixed beds34. Similarly, sonication during carbonation reduced the particle size of the calcium carbonate crystals formed, but it was determined that increased temperature, pressure, and mixing could result in the same conversion over longer periods of time31,32.
The design of aqueous carbonation at a pilot-scale is currently limited by energy and economic costs, particularly because most materials will require pretreatment or significant passivation layer mitigation to achieve high carbon uptakes70. The high energy and economic costs are most often due to pretreatment, chemical additives, separation and disposal of products, and capital installation costs, which would proportionally decrease as scale increases58,75,86.
Indirect carbonation is a multi-step process but must include (1) the leaching of metals of interest from source minerals and (2) the precipitation of carbonates. The leaching step, depending on the feedstock, can compensate for larger particle sizes, competing products such as sulfates, and the formation of certain passivation layers. A general representation of an indirect process, as well as the simplified aqueous chemistry, is shown in Fig. 7. Table 4 gives an overview of indirect processes from the literature.
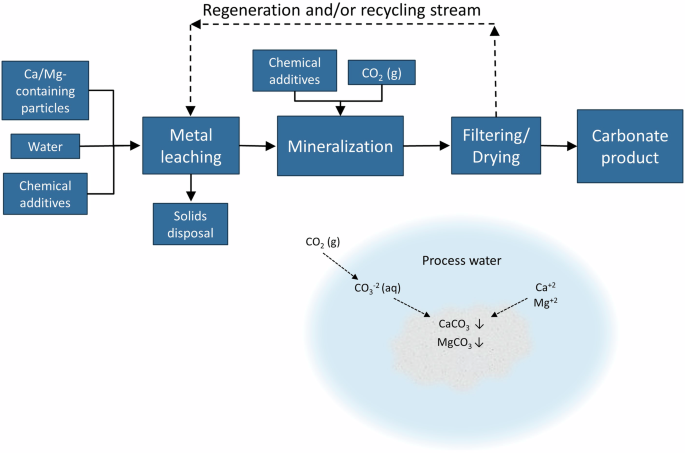
General flow diagram of indirect aqueous processes with carbonation chemistry shown.
Leaching agents are most commonly acids or ammonium salts, determined by the feedstock and desired process conditions26. As a result of the increased solubility of many calcium and magnesium-containing minerals at low pH, many indirect techniques involve some variation of a pH swing during a multi-step process, in which a low pH is used to dissolve metal ions and a high pH is used for carbonate conversion and precipitation.
Leaching techniques used in indirect mineralization include acidic, alkaline, salt-based (such as ammonia salts), complexation, and caustic extraction7,87. Acidic environments can increase the solubility of select elements, such as extracting calcium and magnesium from various oxide phases. However, after acidic leaching, a pH swing is often necessary to return the solution to an alkaline environment where carbonate mineralization is preferred. Ammonia salts are used to increase the solubility of other species in the same aqueous system. Complexation uses additives, typically organics, with carboxyl or hydroxyl groups to dissolve minerals. Figure 8 shows general indirect process diagrams of acid (a) and ammonium salt (b) leaching and carbonation. In general, there is a positive relationship between the concentration of the chemical additive and extraction until an additive-dependent concentration is reached above which diminishing returns are observed88,89.
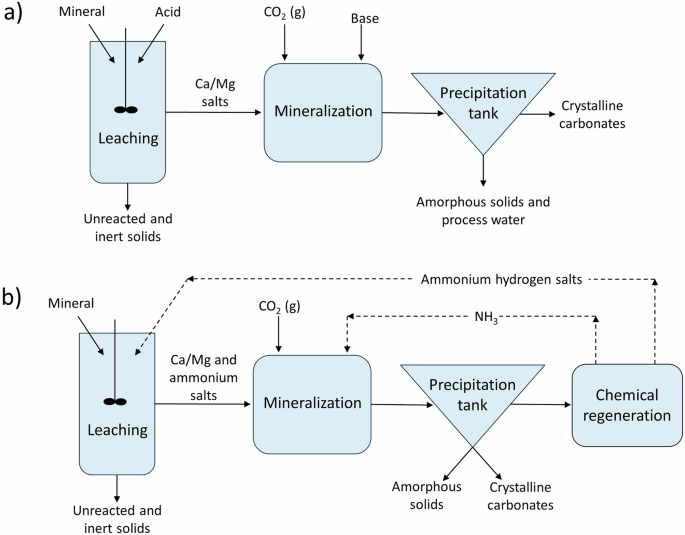
General process flow diagram of a an indirect process using acids to leach metal cations and bases to promote carbonate and b an indirect process using recyclable ammonium salts, ammonium sulfate for example, as a sole chemical additive.
Strong acids like HCl, HNO3, and H2SO4 can extract 80–100% of calcium and magnesium from minerals at concentrations ranging from 0.58 to 4 M26,27,28,30,33,88,89,90,91. These normally correspond to higher stoichiometries than required which improves kinetics but also leaches impurities like iron and silica28,91,92. Selective leaching of calcium has been noticed at lower ratios of strong acids in concrete fines33. The large extraction efficiencies of calcium and magnesium in strong acids indicate leaching is controlled by proton attack
Weak acids and organic additives used for leaching include acetic acid, sulfuric acid, citric acid, ascorbic acid, formic acid, and tributyl phosphate (TBP)26,89,93,94,95. Weak acids often result in incomplete extraction of calcium and magnesium. For example, acetic acid extracted 73% of calcium from paper pulp waste, 75% of calcium from steelmaking slag, and 35% of magnesium from steelmaking slag89,94. Carbonic acid has been shown to selectively leach magnesium from silicates under select conditions14. The addition of TBP aids in the formation of acid-extractant complexes, which can improve the performance of acetic acid94.
Ammonium salts such as ammonium acetate and ammonium chloride can provide pH swing behavior as a single chemical additive and have been used to decrease the cost and chemical requirements of an indirect process57,89,96,97,98. Extraction of alkaline elements by ammonia salts has ranged between 40% and 99% with a direct relationship between concentration and extraction efficiency57,96,97. Selectivity can also be achieved if the chosen salt performs at pHs incompatible with the extraction of other metals57,96.
In general, the consumption of acids and bases during acidic leaching and subsequent pH swings back to alkaline conditions is a costly component of many indirect processes. Some studies using acidic leaching have been able to recycle the acid solution after mineralization/pH swing, but additional research is required to be able to effectively recycle each type of additive for each experiment14,55,89.
The presence of a passivation layer is observed when calcium and magnesium are leached from silicates30. The inert surface layer is predominantly silica but can also contain alumina if it’s significantly present, like in coal fly ash for example57. The passivation layer is responsible for the initial fast extraction rate followed by a plateau in leaching rate30,57. It impedes the diffusion of calcium and magnesium to the particle’s surface and, therefore, to reaction sites28,96. Specifically, the silica phases can form a gel, which can impact the purity of the extraction process but can also be removed through mechanical exfoliation or dissolved and filtered for purification or recovery93,95.
The kinetics of leaching is also influenced by particle size. Decreasing particle size corresponds to improved leaching88,96,97. Several authors have reached high extraction efficiencies at moderate particle sizes (250–425 µm)27,96. Specific surface area, as a replacement parameter for particle size, has also been reported in indirect carbonation99.
Choosing the correct leaching agent for a feedstock is important. Generally, if a feedstock has low calcium and magnesium content or the metals are bound in un-reactive phases, a strong acid is recommended. However, if a process is concerned with impurities, weak acids and organic extractants show significantly less leaching or iron and silica100.
The temperature of the leaching step influences the kinetics and selectivity of leaching processes. Magnesium and iron leaching are more sensitive to temperature change than calcium88. Using elevated temperatures above 90 °C increases the leaching of other species, such as iron, aluminum, and silicon94. Exclusively calcium leached at slightly lower temperatures and shorter reaction times for related materials, indicating that selectivity can be achieved through temperature manipulation97. The separation of leaching and mineralization steps in indirect processes enables independent temperature control of these steps.
In indirect processes using pH swing, NaOH is predominantly used to increase the alkalinity to roughly 9 or 10 and induce carbonate precipitation101. Control of the pH can be used to selectively precipitate either calcium or magnesium carbonates and control the morphology of precipitates. For example, calcite and vaterite were selectively precipitated at pH 1133,91.
The temperature of mineralization can also influence the products and rates89,95,96,97. Several studies concluded an inverse relationship between carbonation efficiency and temperature, due in part to the decreased solubility of dissolved carbon as temperature increases89,96. Generally, higher temperatures favor aragonite formation57.
Similar to direct aqueous mineralization, the mineralization step of indirect processes depends strongly on the partial pressure and dissolution of CO214,89,102. Additionally, the carbonate precipitation step can often be much slower than CO2 dissolution which can upset equilibrium calculations14. A steel slag study concluded that pressure above 30 bar yielded amorphous products which indicates there was likely carbonation of impurities93. Carbonate species, such as ammonium carbonate, have also been chemically added to improve reaction efficiencies57.
Indirect carbonation studies, empirical and modeled, have been carried out or for feedstocks of cement waste, serpentine, and fly ash24,89,92,103,104. One bench-scale study, determined to be carbon negative, had high extraction and carbonation kinetics using atmospheric concentrations of CO2 by bubbling air through the leachate92. One estimate of indirect carbonation of 200 t/yr waste serpentine calculated 0.544 Mt CO2/yr with a 62% process efficiency24.
Indirect processes tend to be more expensive than direct ones because of the increased cost and chemical additive requirements14. However, indirect processes are better suited to control purity and morphology of products, which can increase the economic value of products89. Cost estimates have ranged between $97 and $147/tCO214,105. However, cost-benefit analysis is required on the control and separation of products as applied to actual feedstocks83.
Higher energy requirements of indirect processes factor into the economics and lifecycle emissions of indirect processes89. However, one study determined that their indirect process consumed 300 kWh/t CO2, which is less than estimates for geological sequestration and amine capture14,30,96,106. For example, just 8% ammonia can absorb twice the carbon dioxide as 20% monoethanolamine (MEA) and requires less than 2 GJ/t CO2 as compared to 4 GJ/t CO2106. More generally, barriers for larger scale indirect carbonation include high operating costs, largely due to the price of chemical additives, the low market price of carbon, and incomplete understanding of standards and commercialization of products89,93,103,104.
Among the technologies discussed in this review, several trends arise, as do conflicting and/or dissimilar results. First, temperature has a complex effect on most carbonation reactions because of the decreasing solubility of CO2 at elevated temperatures. The decreased presence of dissolved carbonates at high temperatures in aqueous systems poses a challenge since the reaction between metals and carbonates occurs much more rapidly in solution. Aqueous systems typically have higher carbonation efficiencies, and many have had success at close to ambient temperatures. Additionally, aqueous systems must operate below the boiling point of water. To exceed the carbonate concentration limited by dissolved CO2, some studies have also used ammonium carbonate or bicarbonate with good results. Temperature affects chemical phases present in these industrial waste feedstocks differently, particularly with respect to the process design. Future work can involve exploring the relationship between the activity of each chemical phase and temperature to better understand how to optimize temperature in aqueous systems.
The presence of a passivation layer is constant across mineralization studies but differs slightly between the formation of a product or silica passivation layer. Gas-solid systems where the mineralization reaction occurs in the particle’s pore water form a carbonate product passivation layer that limits further CO2 diffusion to active sites deeper within the particle. Aqueous systems can form both product and silica passivation layers, largely depending on the concentration of carbonate. A nucleation-limited system, with no supersaturation, will more easily form a product passivation layer on the surface of a particle. Additionally, if diffusion kinetics are fast enough to fully dissolve calcium and magnesium from a particle before it mineralizes, distinct product particles can be produced from an aqueous system. The use of the shrinking-core model is dependent on these considerations of the limiting reaction mechanism. Pretreatment is the most common method of passivation layer mitigation, specifically grinding to reduce particle size or during carbonation to remove the passivation layer as it forms. Future work can include low-energy and cost methods of passivation layer mitigation.
The carbonation content of the chosen industrial waste streams can exceed 600 kg CO2/t, but most values are around or below 200 kg CO2/t. Serpentine, with good pretreatment and system design, can have good carbon uptake and efficiencies. This might partially be due to the wealth of research on carbonation serpentine and olivine that doesn’t exist for other phases of calcium and magnesium silicates. The range of carbon uptake for mine tailings in this review is 53–280 kg CO2/t mine tailings. In comparison, steel slag uptake slightly exceeded that range while concrete waste was slightly below those values. The range of carbon uptake for ash waste is 32–635.5 kg CO2/t. These values indicate that operating conditions contribute to considerable differences in carbon uptake but that a certain amount of it is due to material chemistry. For example, concrete waste can contain inert aggregates, but cement paste or kiln dust is far more reactive due to its higher calcium presence. Additionally, calcium and magnesium in steel slag and mine tailings are primarily found in silicate minerals, but ash waste contains different phases of calcium that have different reactivities in mineralization reactions. Something also not considered in this paper is the carbon offset from the use of products from mineralization reactions. For example, many concrete waste studies focus on creating either cementitious materials or aggregate to work towards a circular economy within the cement industry. Replacing Portland cement with mineralized industrial waste indirectly offsets emissions from cement manufacturing which accounts for 8% of global CO2 emissions.
The comparison of cost and energy between processes is valuable for large-scale consideration. First, gas–solid systems are often designed to be cheap because they can operate on large tailings piles using flue gas and require very little equipment. For this reason, they also consume very little energy and, therefore, produce very few CO2 emissions. However, direct gas–solid reactions also have some of the lowest carbon uptakes. Cost estimates of direct aqueous carbonation overlap considerably with indirect but also include many cheaper options per ton of CO2 sequestered. The capital costs of direct and indirect can be similar if a direct process includes precipitation or settling tanks but operating costs consider chemical additives, which indirect processes will normally require more of. Future work for indirect processes can focus on either reducing the volume of chemical additives required or effective recycling. The energy consumption of direct aqueous and indirect processes will be largely determined by operating conditions, but good results have already been achieved at mild conditions which indicates the possibility of mineralization being carbon neutral or negative.
Efforts to bring industrial waste mineralization technologies to pilot-scale largely focus on translating batch reactors to continuous flow using packed or fluidized beds and rotating drums while simultaneously finding reuse potential for the carbonate product, such as lightweight aggregate example. Direct gas–solid systems tend to be the cheapest and least energy intensive, especially at larger scales, since they have very low capital and operating costs. In comparison, direct and indirect aqueous systems are more energy-intensive and expensive because they require more equipment and chemical additives. For this reason, indirect processes have also focused on recycling the acids and bases used for leaching and carbonation, respectively, or using ammonium salts that can provide the required pH swing and be recovered after the carbonation reaction. Future work includes optimizing the storage and transportation of industrial waste to maximize its carbon sequestration potential, testing pilot-scale reactors to determine discrepancies between large and small-scale carbonation, and reducing the requirement for acids and bases in aqueous systems.
Conclusion
Industrial waste streams such as mine tailings, concrete waste, steel slag, and combustion ashes show promising reactivity for carbon mineralization and can add several thousand Mt of sequestration potential to help reach the IPCC goal for reduced carbon emissions. The industrial waste streams focused on in this review have high calcium and magnesium contents, require less pretreatment than natural rock, and are produced in high enough quantities to yield significant carbon sequestration.
Various parameters can be tuned to increase mineralization efficiency. The impact of temperature varies between gas–solid and aqueous systems, where gas–solid processes are able to increase dissolution and diffusion kinetics using hot flue gas (300–600 °C), but aqueous systems are impeded at higher temperatures due to the decreased solubility of carbon dioxide and therefore operate optimally between 25 and 80 °C34,35,55,65,69. Pressure scales more directly with carbon uptake and, combined with temperature, can be manipulated to control product particle size and morphology76,93. In aqueous systems, a high L/S ratio can impede reactant interactions and supersaturation so lower L/S ratios are preferable as long as there is enough water for reactant dissolution. Passivation layers of carbonate product and silica are present in both gas–solid and aqueous systems due to the nucleation of carbonate on the particle surface and leaching of metals from the core of the particle, but both limit further diffusion of reactants and slow reaction rate.

Responses