Systemic lupus erythematosus: updated insights on the pathogenesis, diagnosis, prevention and therapeutics

Introduction
Systemic lupus erythematosus (SLE), canonically defined as an auto-immune disorder, can be considered as a chronic inflammatory illness with clinical manifestations encompassing various organs such as the blood vessels, brain, lungs, skin, kidneys and joints due to polymorphic biological alterations.1 It affects approximately 3.4 million people worldwide, with 400,000 individuals being newly diagnosed each year.2,3 It most commonly occurs among women between puberty and menopause,4 and individuals of the African origin have a higher risk of developing SLE.5,6,7 According to a 2023 global epidemiology study of SLE, Poland, the United States, Barbados, and China showed the highest SLE incidence.2 Though still with an unclear disease of origin, the chance of developing SLE is believed to be associated with genetic factors, epigenetic factors, environmental triggers, and hormonal factors.8
SLE can be diagnosed from the perspectives of disease onset and disease activity. These can be assessed using varied types of evaluation metrics such as the American College of Rheumatology (ACR) criteria and the SLE Disease Activity Index (SLEDAI). Besides, SLE is typically accompanied with increased risks of developing multiple types of comorbidities, with cancer screening being recommended by the European League Against Rheumatism (EULAR)9 and cerebrovascular disease being alarmed among female SLE patients by the American Heart Association.10
SLE is caused by an autoimmune reaction involving both the innate and adaptive immune systems, where an abnormal immune response is directed to nucleic acid-containing cellular particles. The over production of antibodies targeting these nucleic acids, known as antinuclear antibodies (ANAs), is characteristic of SLE.8 Besides, the anti-Smith (anti-Sm) antibody, which is an auto-antibody directed against a component of the spliceosome, is highly specific to SLE, with 20-40% SLE patients versus approximately 1% healthy individuals carrying them.11
Various types of agents have been used for SLE therapeutics, ranging from cytokines, antibodies, hormones, inhibitors, antagonists, to the transfusion of fresh plasma and stem cells. Importantly, while several drugs initially designed for treating other pathological conditions have been shown effective in treating SLE such as the use of the anti-malaria agent hydroxychloroquine as a SLE therapeutic, a plethora of medications have been reported capable of inducing SLE. Examples of this kind include anti-arrhythmic agents such as procainamide, broadspectrum antibiotics such as minocycline, vasodilators such as hydralazine and methyldopa, and antipsychotics such as chlorpromazine. These have unanimously complicated our understandings on the appropriate therapeutics of SLE, rendering it necessary and urgent to delve into the molecular mechanisms driving SLE pathogenesis and classify current therapeutics accordingly by their mechanisms-of-action. This may guide us towards effective management of SLE and, hopefully, help us identify the pivotal signaling axis for the establishment of innovative targeting strategies.
Following the introduction of some basic knowledge of SLE including the history and epidemiology, this review characterized three key determinants of SLE pathogenesis by their mechanisms-of-action, i.e., over-activated immune response, skewed cytokine microenvironment homeostasis, impaired debris clearance machinery; summarized current understandings on SLE diagnosis by disease onset, activity and comorbidity; introduced risk factors predisposing SLE at the genetic, epigenetic, hormonal and entrinsic levels; and classified current SLE preventive strategies and therapeutics by the identified working mechanisms. Our review not only provides comprehensive information on SLE so far available, but also proposes fresh insights on our current understandings of SLE, with a focus on its prevention and therapeutics.
Basics of SLE
Research history of SLE
The history of SLE can be dated back to 400 Before Christ (BC) and divided into three periods, i.e., the classical period, the neoclassical period, and the modern period. The classical period, cornerstoned by Hipocrates who firstly described the possible ulcers of SLE as herpes esthiomenos, identified and documented SLE as a cutaneous disorder.12 The neoclassical period witnessed the manifestations and therapeutics of SLE. During this period, Ferdinand Hebra reported the facial rash associated with SLE as a butterfly rash with illustrations; and Jonathon Hutchison noted the photosensitive nature of SLE, among other milestones. Regarding SLE therapeutics, quinine was firstly used by the Physician Payne, followed by the use of adrenocorticotropic hormone and cortisone by the physician Philip S Hency, and later hydrocortisone by Sulzberger and Witten. In addition, the inductive role of medications such as sulfonamides on SLE was found during this period.12 The modern era was heralded by the discovery of the lupus erythematosus (LE) cell (a bone marrow phenomenon involving the phagocytosis of nuclear material by polymorphonuclear leukocytes) and characterized by rapid scientific advances over the past 60 years. Before the discovery of LE cell by Hargraves in 1948, the Wasserman test for syphilis was used for SLE diagnosis. The use of immunofluorescence for ANA detection followed. The establishment of the murine models has substantially advanced the scientific field related to SLE, leading to the discovery of the genetic predisposition of SLE by Leonhardt and the familial association of SLE by Arnett and Shulman of Johns Hopkins.12 Thanks to the contributions of these scientists made along the history of SLE researches, the lifespan of SLE patients has now been extensively extended from no longer than 5 years after the initial diagnosis to living with illness12 (Fig. 1).

History and etiological features of SLE. The history of SLE studies is divided into three periods, i.e., classical period, neoclassical period, modern period. The classical period is characterized by ‘SLE description’, with critical events being ‘description of SLE as herpes esthiomenos by Hipocrates’, ‘description of SLE as noli me tangere by Rogerius Grugardi & Rolando of Parma’, ‘confusion of SLE with tuberculosis by Erasmus Wilson’, ‘description of SLE as skin disorders by Robert Willan’, and ‘differentiation of SLE from other diseases by Willan’. The neoclassical period is characterized by ‘SLE manifestions, therapeutics, and identification of the inductive role of some medications on SLE’, with representative events being ‘documentation of SLE with illustrations as a butterfly rash by Ferdinand Hebra’, ‘identification of the photosensitive nature of SLE by Jonathon Hutchison’, ‘definition of the two forms of SLE, i.e., discoid and disseminated SLE by Mariz Kaposi’, ‘use of quinine b Payne and use of adrenocorticotropic hormone (ACTH) and cortisone by Philip S. Hency’, ‘use of hydrocortisone by Sulzberger and Witten’, and ‘identificaton of the inductive role of sulfonamides on SLE’. The modern period is characterized by ‘SLE diagnosis and establishment of murine model’, with milestone events being ‘discovery of lupus erythematosus (LE) cell by Hargraves’, ‘use of immunofluorescence for antinuclear antibody (ANA) detection’, ‘use of immunofluorescent microscopy for diagnosis’, and ‘establishment of murine model’, where following murine model establishment, the genetic predisposition and familial occurrence of SLE were consecutively recognized
Epidemiology of SLE
SLE is a heterogeneous disease occurring frequently among women and least common among children.8 SLE most commonly afflicts women between puberty and menopause,13 where the female/male ratio shifts from 3/1 in children to about 9/1 or even 15/1 among adults between puberty and menopause14,15 (Fig. 1).
SLE is associated with an increased risk of premature mortality that has improved over the past 30 years,16 and the risk conveys an ethnic-dependent difference.15,17 The development of lupus nephritis (LN), a SLE-associated renal complication, is considered a strong predictor of an increased mortality risk. SLE patients of African, Chinese and Hispanic origins have shown an increased risk of developing LN18,19 and thus enhanced mortality.18,19,20 Besides mortality, the disease incidence, prevalence, age-of-onset, and morbidity of SLE also vary greatly among regions.17 For instance, the annual incidence and prevalence rates of SLE in the United States each varies from 2 to 7.6 and from 19 to 159 per 100 000 individuals, respectively, for people of different racial backgrounds.5,6 In particular, individuals of the African origin, particularly those who have migrated to America or Europe, exhibit a higher incidence and prevalence, earlier age at the disease-of-onset as compared with those of the north European origin5,6,7 (Fig. 1). Asian have a lower regional risk of developing SLE than people from the United States,21,22 but the prevalence of SLE among people carrying the Chinese background has been reported to be increasing.23 Such ethnic differences among SLE patients may be explained by their different socioeconomic backgrounds, distinct perceptions on the condition, varied risks of getting infection or developing comorbidities especially cerebrovascular diseases, imbalanced availability of the medical resources, and non-uniform adherence to the therapeutics.3,16,17,24
SLE pathogenesis by mechanisms-of-action
SLE is characteristic of increased presentation of autoantibodies such as ANA, anti-Sm, anti-double-stranded DNA (anti-dsDNA) antibody, antiphospholipid (aPL) antibodies, and anti-β2-glycoprotein (aβ2GPI) antibodies,1 which even occur years prior to the clinical onset of SLE.25 Being the primary effectors of SLE inflammation and associated damage,26 autoantibodies form immune complexes (ICs) and deposit on multiple organs such as kidney, skin and central nervous system to induce local inflammation.26,27 Antibodies are produced by plasma cells (PCs) and plasmablasts, the terminally differentiated B cells.28 As the precursors of PCs and important antigen-presenting cells (APCs),29,30 B cells loose tolerance to autoantigens31 and may present them to T cells in SLE patients followed by the activation of Th cells. Stimulated Th cells activate B cells and contribute to their differentiation through clusters of differentiation 40 ligand (CD40L)/CD40 interactions.32 Activated B cells move to germinal centers (GCs) with the help of Tfh cells and follicular dendritic cells (DCs), where B cells generating antibodies with high antigen affinity are expanded and differentiate into the memory B cells (MBC) and antibody-producing PCs.33
Over-production of immunoactivating materials
Interferons (IFNs) are cytokines with pleiotropic roles in immune regulation that can be categorized into type I, II, and III based on sequence homology.34 Type I IFNs represent the largest IFN family comprised of IFNα, IFNβ, IFNω, IFNκ, and IFNε; the type II family includes solely IFNγ; and the type III family contains IFNλ1, IFNλ2, IFNλ3, and IFNλ4.35 Out of the three families of IFNs, type I IFNs play an immunomodulatory role that bridges the gap between the innate and adaptive immune systems.35 That is, the expression of type I IFNs is activated on the trigger of nucleic acids via intracellular pathways such as Toll-like receptor (TLR)-mediated signaling; the binding of type I IFNs to their receptors activates the intracellular signaling cascade involving the Janus kinase-signal transducer and activator of transcription (JAK/STAT) axis that leads to stimulated effectors of the innate and adaptive systems.35,36,37
There has been a well-established positive association between type I IFNs and SLE. Specifically, the blood levels of type I IFNs were documented to be elevated in approximately 50% of SLE patients.38 An even greater percentage of SLE patients were estimated to carry over-represented expression of genes involved in type I IFN-mediated signaling in their peripheral blood cells.39,40
Type I IFNs are generated in response to, primarily, the activation of nucleic acid-binding pattern recognition receptors such as the endosomal TLR3/4/7/9, the cytosolic sensor cyclic guanosine monophosphate-adenosine monophosphate (cGMP-AMP) synthase (cGAS), and the ribonucleic acid (RNA)-sensor retinoic acid-inducible gene (RIG) I like receptors (RLRs)-mitochondreal antiviral signaling protein (MAVS).41 These nucleic-acid sensing pathways are chronically over-activated in many SLE patients, with the pathogenesis roles of TLR7 in SLE being well-established.42 For instance, over-activation of the cGAS-stimulator of IFN genes (STING) pathway has been shown to be crucial in autoimmunity and SLE pathogenesis.40,43
In addition, events altering nucleic acid metabolism may also trigger type I IFNs production, where the essential roles of cytosolic nucleic acid sensors played in SLE have been well characterized.44,45 For instance, ultraviolet (UV) light exposure has been shown capable of enhancing type I IFNs response both locally and systemically, with the evidence being obtained from both the animal model and from the clinics46 (Fig. 2).
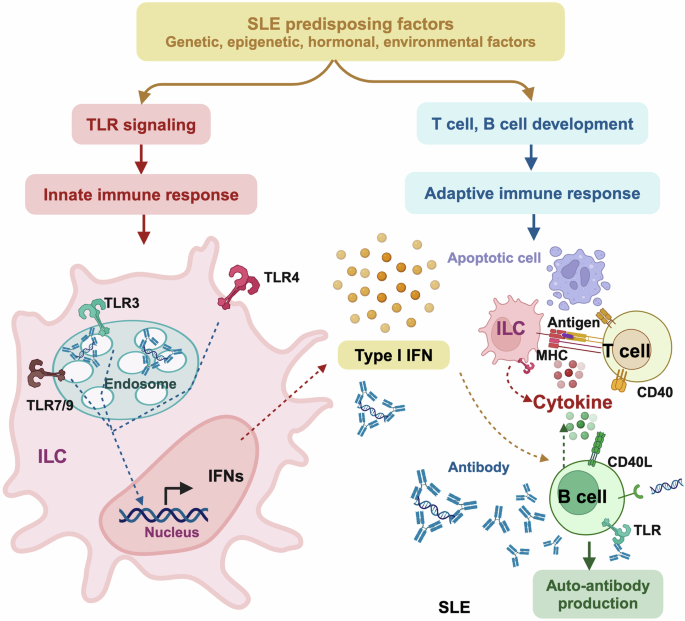
Mechanisms-of-action driving SLE pathogenesis via over-activating immune response. SLE predisposing factors including genetic, epigenetic, hormonal and environmental factors can function as or induce the production of immunoreactants to trigger Toll-like receptor (TLR) signaling in the innate immune system for enhanced generation of type I interferon (IFN), or modulate the development and maturation of T and B cells in the adaptive immune system for over-production of auto-antibodies. Type I IFN functions as the hub bridging the innate and adaptive immune systems. Specifically, type I IFN is produced from innate lymphoid cells (ILC), and directly triggers B cell activation to overtly produce auto-antibodies that ultimately contribute to SLE pathogenesis. Besides producing type I IFNs, ILCs can also present antigens to T cells via major histocompatibility complex (MHC) to activate T cells that express CD40L that interacts with B cells via the CD40L-CD40 bridge for enhanced auto-antibody production
Type I IFNs are central to the activation of both the innate and the adaptive immune systems. Specifically, by interacting with their receptors, type I IFNs induce signaling through the JAK/STAT pathway followed by the transcription of IFNs-responsive genes that encode the ‘IFN signature’ for activated immune response. On the other hand, type I IFNs can directly activate DCs for enhanced presentation of antigens to T cells, where T cell activation and polarization are important to prime B cell differentiation. Activated B cells then produce auto-antibodies that, once over-produced without timely clearance, deposit in organs and cause tissue damages, the process of which is facilitated by the interactions between CD40 and CD40L40 (Fig. 2).
Skewed cytokine microenvironment
There are two types of T cells, i.e., αβ+ and γδ+ T cells, and αβ+ T cells can be further classified into CD4+ and CD8+ T cells. While CD8+ T cells take on the cell-killing activity,47 CD4+ T cells are T helper (Th) cells capable of promoting CD8+ T cell development and B cell differentiation as well as antibody synthesis.48,49 CD4+ T cells can be roughly subdivided into Th1, Th2, Th17 and regulatory T (Treg) cells, based on their distinct cytokine profilings.50 Th1 cells produce, primarily, IFNγ, interleukin (IL)12, IL2, tumor necrosis factor (TNF) alpha, IL1β, granulocyte-macrophage colony-stimulating factor (GM-CSF); Th2 cells secrete IL4, IL5, IL6, IL10, IL13, among others;51 Th17 cells are featured by expressing IL17A, IL17F and IL22;52 and Tregs secrete transforming growth factor beta (TGFβ), IL10, IL34, IL35 etc.53,54 These cytokines form the cytokine microenvironment to support the amplification of a self-directed immune response, perturbed homeostasis of which may lead to disease syndromes including SLE.
With our incremental understandings on the heterogeneity of Th cells and their roles in regulating cytokine homeostasis, more subcohorts of Th cells such as T follicular helper (Tfh), T peripheral helper (Tph), follicular regulatory T (Tfr) cells have been consecutively identified, some of which hold essential roles in SLE.55,56,57,58,59 For instance, expansion of Tfh and Tph is a prominent feature of SLE,58 and Tfr cells can suppress B cell activation via disrupting the recognition and interaction between Tfh cells and B cells.55 B cells have been previously considered to be stimulated by Th2 cells but are now considered primed by Tfh cells. Thus, Tfh and Tph function in the Th2 linkage towards activated B cells and enhanced SLE severity, Tfr cells act as the switch-off button suppressing the activity of Tfh.60 We focus on the Th1/Th2 and Treg/Th17 pairs that build up the conceptual framework controlling cytokine homeostasis, where T cell subsets involved in fine-gained regulations within this framework such as the modulatory roles of Tfh were not comprehensively covered or thoroughly discussed here.
Besides CD4+ and CD8+ T cells, double-negative (DN) T cells (a unique subset of T cells lacking both CD4 and CD8 co-receptors) also play a significant role in SLE pathogenesis. DN T cells are formed by T lymphoid progenitor cells never traveled through thymus, by T lymphoid progenitor cells traveled through thymus but lack further development into CD4+ or CD8+ T cells, and by CD4+ or CD8+ T cells with down-regulated CD4 or CD8 co-receptor.61 DN T cells comprise 1-3% of human T cells and are of high heterogeneity. There exists at least five DN T cell cohorts, i.e., helper DN, cytotoxic DN, innate DN, resting DN, and intermediate DN.62 DN T cells may be pro-inflammatory such as IL17-producing DN T cells63 and anti-inflammatory such as IL10-producing DN T cells.64 The roles so far reported on DN T cells in SLE are largely pro-inflammatory. Specifically, the amount of DN T cells was considered to be positively correlated with SLE activity;65,66 DN T cells conveyed a positive impact on B cell-mediated antibody production by stimulating the release of IL4, IL17 and IFNγ;67 and enhanced DN T cell apoptosis as a result of inhibited neddylation attenuated lupus progression in murine models.66 Though the significance of DN T cells in SLE pathogenesis is not negligible, they are exempted from the focus of this section given the shared cytokines (such as IL4, IL17 and IFNγ) they produce with different CD4+ T subsets, complex functional plasticity (having both helper and cytotoxic DN cohorts) and small percentage.
Th1/Th2 imbalance
Cytokines produced by Th1 cells are largely pro-inflammatory, and those generated by Th2 cells are primarily anti-inflammatory.68 Numerous evidence has suggested that abnormal T cell differentiation to Th2 dominance can lead to B cell hyper-activation that contributes to immune disorders including SLE pathogenesis.51 The selective development of Th1 and Th2 cells is primarily driven by the cytokine microenvironment among other influential factors such as antigen dose, affinity of antigens, major histocompatibility complex (MHC) haplotypes and co-stimulatory factors. Among other cytokines, IL12 and IL4 dictate the fate of Th cells to the Th1 or Th2 linkage, respectively. While IL12 drives Th1 cell differentiation through STAT4 signaling that leads to up-regulated IFNγ and down-regulated IL4/IL5 for amplified Th1 proliferation, IL4 induces Th2 clonal expansion through STAT6 that results in up-regulated levels of IL4/IL5 and down-regulated IFNγ expression for augmented Th2 differentiation (Fig. 3).69
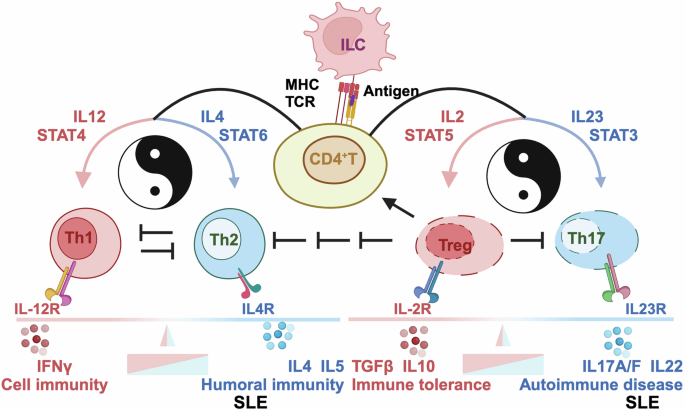
Mechanisms-of-action driving SLE pathogenesis via skewing cytokine microenvironment. On antigen presentation to CD4 + T cells through T cell receptor (TCR) by innate lymphoic cells (ILC) via major histocompatibility complex (MHC) from the innate immune system, CD4+ T cells are activated and can differentiate into distinct T cell subsets such as T helper 1 (Th1), T helper 2 (Th2), T helper 17 (Th17) and T regulatory (Treg) cells, the direction of which is dictated by the cytokine milieu of the microenvironment. Specifically, when IL12 is enriched in the environment, T cells favor Th1 polarization, the process of which involves STAT4 signaling that leads to up-regulated IFNγ and down-regulated IL4/5; when IL4 is enriched in the milieu, T cells are triggered for Th2 clonal expansion via STAT6, resulting in up-regulated IL4/5 expression and down-regulated IFNγ level. On the other hand, IL23, among others, drives T cell plasticity towards Th17 phenotype, the process of which involves STAT3 signaling; and IL2, out of other cytokines, inhibits Th17 generation but promotes Treg generation, where STAT5 plays a role. The balances between Th1 and Th2 cells dictates the preference towards cell immunity and humoral immunity, respectively, as regulated by IFNɤ and IL4/5, respectively. The homeostasis between Treg and Th17 cells determines whether the system goes for immune tolerance (that can cause chronic infectious diseases including cancers) or immune activation (that can lead to the pathogenesis of autoimmune diseases including SLE), as regulated by TGFβ, IL10 (among other cytokines produced by Treg cells) and by IL17A, IL17F, IL22 (out of other cytokines generated by Th17 cells). These subsets of CD4+ T cells cross-regulate among themselves. In particular, Th1 and Th2 cells suppress the expression of each other, Treg cells reduce the levels of Th1, Th2, Th17 cells, and activates that of CD4+ T cells. Only representative cytokines are listed in this Figure. that do not exclude the existence of others
Th17/Treg imbalance
The imbalance between pro-inflammatory Th17 cells and immuno-suppressive Tregs underlies the pathogenesis of SLE.70,71,72 The proportion of Th17 cells is higher in SLE patients, the content of which is positively correlated with SLE severity.73 Tregs play an important role in maintaining the immune tolerance, reduced levels or activities of which are tightly associated with the onset and progression of SLE74,75,76 (Fig. 3).
The subsets of Th17 cells and Tregs have distinct metabolic patterns. While Th17 cells are mainly powered by glycolysis, the energy supply of Tregs largely relies on fatty acid oxidation and oxidative phosphorylation.77 Accordingly, glycolysis deprivation was found to impair Th17 cell differentiation but support the growth of Tregs;78,79,80,81 and impaired fatty acid oxidation was associated with increased Th7 cell linkage yet diminished Tregs development.82 Thus, switching energy supply from relying on carbohydrates to lipids (low-carb or ketogenic-diet) may be a dietary recommendation for SLE patients.
Impaired debris clearance machinery
The pathogenesis of SLE is associated with the failure of removing self-reactive clones of T and B cells. Under normal conditions, an immune response against self-antigens (i.e., anergic responses) can be suppressed by the immune system; however, when this debris clearance machinery is impaired, the ICs (comprised of, e.g., nucleic acids, nucleic acid-binding proteins, autoantibodies directed against those components) may form and initiate the onset of inflammation and organ damage; perpetuation of damage occurs when the ICs further amplifies the immune system followed by the trigger of downstream signals that induce pro-inflammatory mediators such as IFNα, leading to or aggregating the pathogenesis of SLE.
The complement system is centered at the core of the immune system mediating a cross-talk between the innate and adaptive immune responses. The complement system has been implicated in diverse biological processes in mammals including, e.g., modulation of the immune tolerance, and autoimmune diseases. Dynamic homeostasis between the activation and inhibition of the complement system is required to maintain human health. While hyper-activated complement system may lead to excessive inflammation and tissue damage, hypo-activation of the complement machinery may impair debris clearance and lead to autoimmune disorders including SLE83 (Fig. 4).
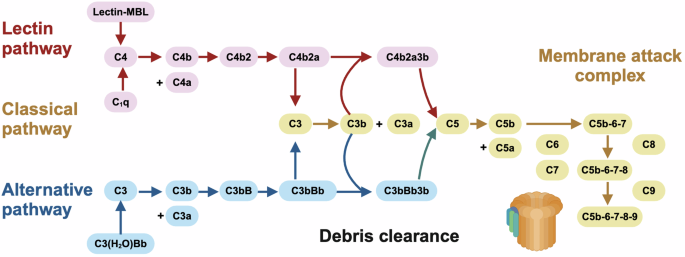
Mechanisms-of-action driving SLE pathogenesis via impairing debris clearance machinery. There are three mechanisms to initiate the complement cascade for debris clearance, i.e., the classical, lectin, and alternative pathways. In the classical and lectin pathways, the complement system is triggered by the binding of the antibody complexes to C1q of the C1 complex and the binding of foreign carbohydrate moieties to mannose binding lectin (MBL) or ficolin, respectively, which converge to the cleavage of C4 to C4b and C4a followed by the generation of one C3 convertase, i.e., C4b2a. In the alternative path, the complement system is activated via spontaneous hydrolysis of C3 into C3b and C3a by the convertase C3(H2O)Bb followed by the formation of another C3 convertase, i.e., C3bBb. Under the cleavage of C4b2a or C3bBb, C3 becomes C3b and C3a. Following this, C3b binds to C4b2a or C3bBb to form C4b2a3b or C3bBbC3b which are C5 convertases capable of hydrolyzing C5 into C5b and C5a; and C5b can initiate the cascade of forming the membrane attack complex (MAC) capable of generating pores in the membranes of pathogens or targeted cells. While C3 activation fragments such as C3b participate in the cleaning of cellular debris to avoid overt activation of the immune system that is favorable for halting SLE pathogenesis, overt production of MAC may promote SLE pathogenesis via causing cell death and generating more immunoreactants
The complement cascade is activated to initiate the proteolytic cleavage of complement proteins into fragments that relay signals to neighboring cells and leukocytes by being deposited onto the targets or released into the extracellular fluid. There are three mechanisms, so far elucidated, to activate the complement system, i.e., the classical, alternative, and lectin pathways.83 In the classical pathway, the complement system is triggered by the binding of the antibody complexes to C1q of the C1 complex. In the alternative path, the complement system is activated via spontaneous hydrolysis of C3. In the lectin pathway, the system is stimulated via the binding of foreign carbohydrate moieties to mannose binding lectin (MBL) or ficolin. All pathways converge to the generation of C4b2a or C3bBb, the cleavage of C3, and the amplification loop. C3b, cleaved from C3, then promotes the formation of C4b2a3b in the classical/lectin pathways or C3bBb3b in the alternative pathway. C4b2a3b or C3bBb3b activate C5 to form the membrane attack complex (MAC) that takes on the action through direct lysis of the target cells. Perturbed activation or availability of any part of this cascade may impair immune homeostasis and lead to severe clinical syndromes such as SLE. For instance, over-expression of complement C3 has been associated with promoted gastric cancer progression via activating JAK2/STAT3 signaling84 (Fig. 4).
Besides the complement system, other mechanisms also exist for debris clearance. For instance, marginal zone macrophages (MZMs) are crucial for clearing apoptotic cells and maintaining immune tolerance, dysfunction of which can lead to defected clearance of apoptotic cells, activated autoreactive T cells and expanded DN T cells.63
Diagnosis of SLE
Disease onset
SLE is highly heterogeneous with variable distinct clinical manifestations, and the disease severity varies from mild to moderate and to severe. For instance, skin inflammation might be restricted to malar rash in one individual but involve upper extremity and trunk as well in another.
The diagnosis of SLE is challenging as no consensus has been made on the diagnostic criteria that needs to be of both a high specificity and a high sensitivity.85 Currently, SLE is diagnosed by both clinical manifestations and laboratory examinations, where SLE manifestations can be defined by the presence of both subjective and objective findings, as well as laboratory examinations. Subjective observations include, e.g., headaches, chest pains, and arthralgias. Objective documentations include, e.g., electrocardiographic or echocardiographic confirmation of cardiac comorbidities. Lab tests include, e.g., autoantibody detection, functional test and imaging.8
The 1997-version ACR classification criterion has been canonically used for SLE diagnosis,86,87 with the classification indexes used being malar rash, discoid rash, photosensitivity, oral ulcers, non-erosive arthritis, pleuritis or pericarditis, renal disorder, neurological disorder, hematological syndrome, immunological evidence, and positive ANA. In the clinical practice, malar rash refers to erythema over the malar eminences that tends to spare the nasolabial folds; discoid rash is defined as erythematous raised patches with adherent keratotic scaling and follicular plugging; photosensitivity is documented as skin rash on sunlight exposure; oral ulcers is considered as oral or nasopharyngeal ulceration that is typically painless; non-erosive arthritis is defined as tenderness, swelling or effusion in peripheral joints; pleuritis or pericarditis refers to rubbing or evidence of pleural/pericardial effusion; renal disorder is considered if persistent proteinuria of >0.5 g/day occurred or cellular casts were present in urine including red blood cells or hemoglobin; neurological disorder primarily refers to seizures or psychosis; hematological disorder refers to hemolytic anemia with reticulocytosis, leukocytopaenia, lymphocytopaenia, or thrombocytopaenia; immunological evidence refers to the presence of anti-deoxyribonucleic acid (DNA) autoantibody, anti-Sm autoantibody, or aPL autoantibodies; and positive ANA is defined as abnormal presence of ANA.8 These 11 indexes were updated in the 2019 EULAR/ACR classification criterion, with ‘fever’, ‘autoimmune hemolysis’, ‘non-scarring alopecia’, ‘low complement levels of C3 and/or C4’ being included, and ‘malar rash’ and ‘photosensitivity’ being removed. In addition, positive ANA was considered as an entry criterion for SLE characterization and a weighted system was used to assess the clinical and immunological manifestations of SLE in the 2019 EULAR/ACR classification criterion.88,89 Though specificities of these two ACR versions remain similar (i.e., 93%), the sensitivity of the 2019-version improved from 83% to 96% as compared with the 1997-version88 (Table 1).
The ACR criteria have been considered to be more feasible for classifying advanced SLE patients. This is because that the 1997-version requires the presence of no less than four items and the 2019-version requires even more indexes, as well as the fact that the symptoms accrue as the disease progresses.87,88,90
It is worth noting that the ACR criteria include the most prevalent manifestations but not all. For instance, the mucocutaneous manifestations included in the ACR criteria focus on discoid lupus and oral ulcers that do not cover other skin symptoms such as subacute cutaneous lupus, psoriasiform, and other forms of chronic cutaneous lupus; clinical syndromes from the neurological system is poorly represented in both the 1997 and 2019 versions of the ACR criteria that lack other important manifestations such as organic brain syndrome and cerebrovascular accident.91 Other SLE classification criteria also exist such as the Systemic Lupus International Collaborating Clinics (SLICC) criterion that overcomes issues faced by the ACR criterion such as the lack of cutaneous and neuropsychiatric manifestations92 (Table 1). However, the specificity of the SLICC criterion drops from 93% to 84% despite its comparable sensitivity (i.e., 97%) with the 2019-version ACR criteria (i.e., 96%) due to the large spectrum of SLE manifestations it includes.87,92 Also, the SLICC classification criteria did not make substantial improvement in diagnosing patients with early disease onset as compared with the ACR criterion. Thus, unless more clever criteria with both increased sensitivity and specificity could be developed, the 2019-version ACR criterion may remain prevalent for SLE diagnosis. However, to enable accurate diagnosis of SLE with only a few indexes is challenging due to the extreme heterogeneous nature of this disease regarding its diversified clinical manifestations. One possibility would be to use molecular markers that requires in-depth understandings on SLE pathogenesis and identification of the leading signaling axis or panel of molecules marking the initiation and/or progression of SLE. Specifically, stratifying the pathogenic process of SLE into vital stages and identifying markers characterizing each phase may clearly mark the disease cause and activity of an individual on diagnosis. This may not only aid in the therapeutic design for precision medicine despite the heterogeneity nature of this disease, but also enable early diagnosis as markers are grouped by the stages following the line of disease initiation and progression.
Disease activity
Assessment of SLE activity conveys significant clinical values as it is a prognostic factor associated with mortality.93 However, accurate SLE activity measure is challenging due to the multifaceted clinical manifestations SLE possess and its extreme variation features over time.
Multiple indexes have been developed to assess the activity of SLE from multiple dimensions. The Systemic Lupus Activity Measure (SLAM) is a scale developed to assess the disease activity of SLE, which comprises items across 11 organ systems but does not require immunological test results and can be scored based solely on the physician’s clinical examination, making it applicable in areas where laboratory testing is limited. SLAM has a relatively satisfying high sensitivity and is currently widely used around the world.94 The SLEDAI95 together with its updated versions85,91,96 have been used to describe the overall burden of SLE. The Adjusted Mean SLEDAI-2000 (AMS) has been developed to measure the disease activity over time.97 However, SLEDAI and it updated versions have limitations in detecting clinically meaningful changes in the disease activity, as only complete remission but not partial improvement of the disease status can be captured using SLEDAI. Thus, SLE disease activity score (SLE-DAS) has been established accordingly to overcome such obstacles, which has displayed a desirable sensitivity for assessing alterations in disease activity.98 The British Isles Lupus Assessment Group (BILAG) criteria and revisions99,100,101 are organ-specific indices assessing the partial improvement of SLE activity that can be used alone or as part of a composite index such as the BILAG-based Composite Lupus Assessment (BICLA).102 BICLA is integrated from BILAG, SLEDAI and Physician Global Assessment (PGA),103,104 which can comprehensively evaluate the benefits of an individual patient from a particular therapeutic towards an efficient utilization of the medical resources,105 and thus has been adopted by several clinical trials.106,107,108,109,110 Similarly, the SLEDAI-2000 Responder Index-50111 and composite indices such as the SLE Responder Index (SRI)112 are also available for assessing partial SLE improvement. The SLICC ACR Damage Index (SDI) has been developed to measure the accumulated organ damage ever since the disease onset,113 which has been shown to be a reliable114 independent outcome measure115 as well as a predictive index on future damage accrual and mortality.116 In addition, lupus low disease activity (LLDAS) has been used to describe prolonged disease remission or a serologically active (i.e., high anti-dsDNA antibody or low levels of complements) but clinically quiescent period among SLE patients.117 Therapeutically, patients during the LLDAS phase do not need specific treatment but require close surveillance118,119 (Table 1).
Disease comorbidities
As a direct result of SLE or a consequence of SLE medications such as glucocorticoids, SLE patients are at a high risk of developing several comorbidities. Thus, surveillance using measurements of each type of comorbidity should be adopted to prevent SLE patients from developing these diseases, which is crucial to their early identification and the prevention/intervention of these SLE-associated disorders among these patients.
Primary SLE comorbidities include cancers such as hematological malignancies, cardiovascular disorders such as atherosclerosis, bone diseases such as osteonecrosis, and neuropsychiatric symptoms such as cognitive dysfunction. In particular, SLE is associated with an increased risk of developing cancers, especially breast cancers, cervical cancers, hematological cancers, and lung cancers,120 with cancer screening being recommended for SLE carriers by the EULAR.9 SLE has been considered as a risk factor for atherosclerosis and been incorporated into the American Heart Association guidelines for cerebrovascular disease prevention among women.10 In addition, coronary artery disease was documented in 6-11% SLE patients, and subclinical carotid plaque was reported in 30-50% SLE carriers.121 SLE patients are at a high risk of developing osteonecrosis and osteopenia. Specifically, the incidences of osteoporosis and osteopenia occur in 1.4-68% and 25-74% SLE patients, respectively.122 Besides, SLE-related factors such as disease activity and medication use have been reported to be risky for developing low bone mineral density.122 Cognitive impairment occurs in up to 88% neuropsychiatric SLE123 that requires early diagnosis and appropriate interventions to prevent its long-term damage accumulation. Thus, there is an urgent need of standardized metrics for identifying cognitive impairment that, however, is lacking.124
Risk factors of SLE
Risk factors of SLE can be classified into intrinsic and extrinsic levels, with intrinsic factors being further grouped into those occurring at the genetic, epigenetic and hormonal levels, and extrinsic factors classifiable into environmental factors, habits, physiological factors and phycological factors. Before going into details of each type of SLE risk factors, it is worth to note that factors predisposing the risk of developing SLE is multifactorial that can not be explained solely by information from any of these layers, and risk factors from multiple levels can synergize to predispose the onset and severity of SLE. As examples, increased risks of developing SLE have been observed when genetic risk factors interact with smoking,125 and when the risk allele of the gene encoding chymotrypsin-like elastase family A member 1 (CTYP24A1) is coupled with insufficient vitamin D supply.126
While we focus more on intrinsic factors in this section and go into details of those at the genetic, epigenetic and hormonal levels to gain insights for intrinsic SLE predisposition, we emphasize extrinsic factors in the following section to identify preventive approaches for practical advice.
Genetic factors predisposing SLE
SLE is an autoimmune disease with a strong genetic disposition. Approximately 5-12% of people having one of his/her first-degree relatives carrying SLE will develop this disease in their lifetime.127 A series of landmark familial linkage studies and genome-wide association studies (GWAS) in SLE have greatly advanced our understanding regarding the genetic basis of SLE.128,129,130 Currently, more than 100 SLE susceptibility loci have been consecutively identified (mostly from the European and Asian populations), which can explain up to 30% of SLE inheritability.131,132,133,134
Being a multigenic disease, several weighted genetic risk scores (GRS) have been established to assess the cumulative genetic susceptibility of an individual to SLE,125 with a higher GRS being associated with an earlier SLE onset and a higher disease activity.135 Male SLE patients, in general, have higher GRS than female carriers, implicating that the genetic factors play more dominant roles among males than females in predisposing SLE susceptibility.136
Genetic factors predisposing the incidence of SLE include both high-risk rare mutations and high-frequency polymorphisms (SNPs) that aggregate to collectively enhance the susceptibility of an individual to SLE. Of note, though each conveying a small effect on SLE risk by itself, low-frequency SNPs, once aggregated in a sufficient amount, may deliver substantial impact on SLE susceptibility.137 Genetic alleles so far identified are largely distributed in genes participating in IFN-relayed signal transduction, genes encoding components of the MHC region and altering the threshold for activating T/B lymphocytes, and genes encoding elements of the complement system such as C2, C4, C1q that impair the clearance of cellular debris.129,137,138,139 Genetic factors predisposing SLE susceptibility can be classified into three categories according to the molecular mechanisms they may participate in, i.e., stimulating the immune system, skewing immune regulatory signals, and impairing the debris clearance machinery.
Genetic factors associated with stimulated immune response
A large number of SLE-associated SNPs have been mapped to genes encoding proteins regulating or in response to type I IFNs such as genetic variants of IFN regulatory factor (IRF) 5 and IRF7.140 These SLE-associated genes are known as the ‘IFN signature’ actively participating in the innate immune response, and SLE patients possessing high levels of IFNα are inclined to manifest more severe disease syndromes.141 Mechanically, type I IFNs are produced in response to foreign material invasion for promoted maturation of DCs and production of proinflammatory cytokines, leading to, e.g., stimulated Th1 polarization and B cell activation. IRF5, being one member of the IFN signature with critical roles in regulating type I IFN-responsive genes, conveys a modest contribution to SLE risk (with the odds ratio being 1.5) and is considered to be the most strongly SLE-associated gene outside of MHC.142 As one example of SNPs of this kind, rs12537284 was identified through GWAS followed by meta-analysis and candidate gene investigations.143 STAT4 has recognized as a susceptibility gene of SLE that carries an additive value with IRF5 for increased risk of developing SLE.144 Accordingly, SNP risk variants rs3821236, rs3024866, rs7574865, being associated with high levels of STAT4 expression, conferred increased sensitivity to IFNα signaling in the peripheral blood mononuclear cells (PBMCs) of SLE patients and displayed earlier disease onset and more severe disease syndromes.145 Besides IRF5, SNPs associated with genes encoding other type I IFNs such as rs4963128 of IRF7146 and rs116440334 of IRF8147 have also been implicated in IFN pathways. Several other genes have also been identified capable of influencing IFNα signaling and the innate immune response. These include, e.g., IRAK1 (encoding IL1 receptor-associated kinase 1) that can be used to explain the female-predominance feature of SLE,148 and OPN (encoding osteopontin) that is associated with early SLE onset.149
In addition, SNPs residing in the genetic elements of microRNA (miRNA) critical for relaying IFN signals have been characterized.150 For instance, rs57095329, located in the promoter region of miRNA-146a, has been found to be highly associated with SLE susceptibility.150 Specifically, individuals carrying the risky G allele exhibited significantly reduced level of miRNA-146a than those carrying the protective C allele; and this may be attributed to the altered binding affinity of the transcription factor (TF) ETS proto-oncogene 1 (Ets-1) to the promoter region of miRNA-146a as a result of this genetic polymorphism.150
Genetic factors associated with immune signal relay
Another important portfolio of genetic risk factors predisposing SLE development are located in genes associated with the MHC especially human leukocyte antigen (HLA)-DRB1 in the MHC class II region.151 HLA molecules play vital roles in auto-antibody production, as risk residues associated with the production of characteristic auto-antibodies (i.e., DRB1 residues 11, 13, 30) being located in the peptide-binding groove of HLA-DRB1.152 Furthermore, HLA-DRB1 and HLA-DQB1 form the most significant haplotype, and seven residues (i.e., HLA-DRB1 residues 13, 11, 37, HLA-DQB1 residue 37, HLA-DPB1 residue 35, HLA-A residue 70, and HLA-B residue 9) collectively increase the explainable heritability of SLE due to HLA to 2.6%.152 Many other genes with essential roles in relaying signals downstream of activated T and B cell surface antigen receptors in the adaptive immune response and auto-antibody production have also been linked to SLE susceptibility.153,154 For instance, risky alleles of SNPs rs2230926 (TNFAIP3 that encodes TNFα induced protein 3),155 rs2476601 (PTPN22 that encodes protein tyrosine phosphatase non-receptor type 22),156,157 rs7829816 (LYN that encodes lymphocyte-specific protein tyrosine kinase),158 rs10513487 (BANK that encodes B cell scaffold protein with ankyrin repeats),153 rs7812879 (BLK that encodes B-cell receptor-associated protein kinase),159 rs340630 (AFF1 that encodes AF4/FMR2 family member 1),160 rs4810485 (CD40),161 rs3433034 (CSK that encodes C-terminal Src kinase),162 rs17849502 (NCF2 that encodes neutrophil cytosolic factor 2),163 rs1057233 (PU.1 that encodes purine rich box-1)164 have been reported to alter T and/or B cell activation threshold in the adaptive immune response for enhanced chance of developing SLE.
Genetic factors associated with debris clearance ability
As the classical pathway activating complement signaling may help remove apoptotic and damaged cells as well as ICs for reduced risk of developing autoimmunity to nuclear components,165 low copy numbers of C4 and C1q or deficiency of these genes are typically associated with increased SLE incidence. It has been estimated that the chance of developing SLE among individuals harboring congenital genetic deficiencies of C4 increased up to 90%.127 Several genetic mutations associated with the complement genes have been documented. For example, the C4A-null allele was associated with a doubled SLE susceptibility than either HLA-B8 or HLA-DR3,166 where HLA-B8 and HLA-DR3 alleles were known to predispose the risk of SLE via influencing early stages of adaptive immune activation.167 C1qA gene deficient mice have a greater chance of accumulating apoptotic bodies in the kidney and developing auto-antibodies to nuclear antigens.168 Also, C1q was shown protective against SLE via directing the immune stimulatory complexes to monocytes rather than DCs that secrete the pro-inflammatory cytokine IFNα,169 and through modulating CD8+ T cell mitochondrial metabolism for reduced immune response to self-antigens.170 In addition, multiple high-frequency low-risk genetic polymorphisms residing in 1q36 have been linked to genes encoding components of the complement system and been considered responsible for debris clearance.171,172,173
Epigenetic factors predisposing SLE
Epigenetic factors predisposing SLE include, primarily, miRNA, DNA methylation, and histone modification.
miRNA
miRNAs can act similarly as the TFs or interplay with TFs to cooperatively regulate the expression of target genes.174 Numerous studies have demonstrated the biological and clinical relevance of miRNAs in SLE. For instance, 42 differentially expressed miRNAs were identified from the PBMCs of SLE patients, among which 7 miRNAs (i.e., miRNA-10a, miRNA-130b, miRNA-134, miRNA-146a, miRNA-31, miRNA-95, miRNA-99a) were more than 6 folds lower in the diseased group as compared with the control.175 4 miRNAs (i.e., miRNA-371-5p, miRNA-423-5p, miRNA-638, miRNA-663) and 1 miRNA (i.e., miRNA-1224-3p) were found to be up- and down-ward regulated, respectively, in LN cells from a study investigating the miRNA profiles of Epstein-Barr virus (EBV)-infected B cells and frozen PMBCs from LN patients and unafflicted controls of different racial groups (i.e., American of African and European origins).176 While being up-regulated in this study, miRNA-423 and miRNA-663 were reported to be down-regulated in another study examining the miRNA profiles of kidney biopsy specimen, where 66 miRNAs were identified differentially expressed in LN cells.177 Focusing on T and B lymphocytes, it was reported that 3 miRNAs (i.e., miRNA-21, miRNA-25, miRNA-106b) were up-regulated in both T and B cells of SLE patients, 8 miRNAs (i.e., let-7a, let-7d, let-7g, miRNA-148a, miRNA-148b, miRNA-196a, miRNA-296, miRNA-324-3p) and 4 miRNAs (i.e., miRNA-15a, miRNA-16, miRNA-150, miRNA-155) showed altered expression in solely T and B cells of SLE patients, respectively, from a study analyzing the expression of 365 miRNAs in PBMCs from 34 SLE patients and 20 healthy individuals.178 Another group reported 11 miRNAs differentially expressed in CD4+ T cells from SLE patients, out of which 6 (i.e., miRNA-1246, miRNA-126, miRNA-1308, miRNA-574-5p, miRNA-638, miRNA-7) were up-regulated and 5 (i.e., miRNA-142-3p, miRNA-142-59, miRNA-155, miRNA-197, miRNA-31) were down-regulated.179 Although numerous miRNAs have been found dysregulated in human SLE patients or pre-clinical animal models, quite few miRNAs and their altered profilings were overlapping across studies, some of which even showed inconsistent patterns. This can be, at least, partially explained by the diversified manifestations and activities of SLE, as well as its heterogeneity regarding, e.g., ethical race and medication history. This makes miRNAs showing conserved alteration profiles among SLE patients highly valuable from the perspectives of both diagnosis and therapeutics.
miRNA-155: promoting SLE
Multiple lines of evidence have indicated that miRNA-155 is activated in response to the stimulation of TLR ligands,180,181 and up-regulated miRNA-155 is associated with activated TLR signaling. Multiple targets of miRNA-155 have been shown with critical roles during TLR signaling. For instance, suppressor of cytokine signaling (SOCS1), being a target of miRNA-155, participated in IFN-mediated antiviral response and thus attenuated viral propagation in macrophages.182 Inositol polyphosphate-5-phosphatase D (INPP5D), another target of miRNA-155, negatively regulated TLR4 signaling in response to lipopolysaccharide (LPS) stimulation.183 Myeloid differentiation primary response protein 88 (MyD88), being targeted by miRNA-155, acted as a vital adapter molecule in TLR signaling.184 TGF-β-activated kinase 1 (TAK1)-binding protein 2 (TAB2) is a direct target of miRNA-155 that activated TLR-mediated nuclear factor kappa B (NF-κB) in LPS-activated DCs185 and plasmacytoid DCs.186 Besides, miRNA-155 played an essential role in dictating the antigen-presenting activity of DCs by suppressing the expression of PU.1 and cellular oncogene fos (c-Fos), two TFs with critical functionalities in regulating DC maturation,187 and defect DCs were associated with attenuated immune response.188,189
miRNA-155 also modulates the cytokine microenvironment by altering the distribution profiles between Th1 and Th2 cells. Specifically, altered Th1 function, skewed Th2 differentiation, and defective B-cell class switching were observed in mice carrying miRNA-155 deficiency as a result of abnormal secretion of cytokines such as TNFα, IL4 and IL10, and IFNγ.190,191 In addition, miRNA-155 affects cytokine homeostasis by changing the distribution between Th17 and Treg cells. For instance, miRNA-155 was negatively involved in Treg cell-mediated tolerance, as miRNA-155 depletion resulted in enhanced Treg-mediated immune suppression;192 and miRNA-155 knockout mice exhibited mass loss of Th17 cells coupled with marked reduction of inflammatory Th17 cytokines.193
miRNA-146a: suppressing SLE
On the opposite to miRNA-155, miRNA-146a level was negatively associated with the risk of SLE,194 substantially down-regulated in SLE patients, and adversely correlated with SLE activity.195,196
miRNA-146a functions as a strong negative regulator of TLR signaling by repressing TNF receptor-associated factor 6 (TRAF6) and IRAK1.197 miRNA-146a expression was inversely correlated with TNFα production, rendering cells tolerant and cross-tolerant to TLR stimulus.198,199 In line with these, miRNA-146a was found capable of regulating the production of type I IFNs (i.e., IFNα, IFN-β).175 Specifically, in SLE patients lack of miRNA-146a expression, aberrant accumulation of the targeted proteins of miRNA-146a (such as STAT1, IRF5, TRAF6, and IRAK1) led to altered activation of the IFN pathway;175 and exogenously introducing miRNA-146a into PBMCs from SLE patients dramatically alleviated the overtly activated type I IFN signaling.175 The latter can be evidenced by the approximately 75% reduction on the transcriptional levels of 3 IFN-inducible genes, i.e., IFN-induced protein with tetratricopeptide repeats 3 (IFIT3), myxovirus resistance 1 (MX1), and 2′,5′-oligoadenylate synthetase 1 (OAS1).175
Similar to miRNA-155, miRNA-146a regulates the cytokine microenvironment by affecting both the Th1/Th2 and Th17/Treg balances. Specifically, miRNA-146a was highly expressed in Treg cells in response to T cell receptor (TCR) activation, leading to impaired IFNγ-dependent Th1 activity and IL2 secretion, the process of which involved STAT1.200,201
Other miRNAs
miRNAs modulating immune response
Numerous evidence has supported the notion that miRNAs are essential players in TLR signaling for stimulating immune response. For example, miRNA-124 suppressed macrophage activation by repressing the expression of CCAAT/enhancer-binding protein alpha (C/EBPα),202 miRNA-126 reduced the expression of PU.1 and thus TLR activation in allergic asthma.203 It is noteworthy that the same miRNAs may not act uniformly under distinct cellular contexts or pathologic conditions. For instance, let-7i, capable of negatively regulating TLR4 expression, was down-regulated in human cholangiocytes204 but up-regulated in DCs in response to LPS stimulation.205
miRNAs play critical roles in the regulation of T cell development. For example, miRNA-184 restricted the activation of CD4+ T cells during the early adaptive immune response and thus limited the production of IL2 by targeting nuclear factor of activated T cells 1 (NFAT1);206 miRNA-181c showed a similar function, the ectopic expression of which suppressed IL2 expression and thus reduced the proliferation of activated CD4+ T cells;207 furthermore, IL2 induced the expression of miRNA-182, leading to inhibited activity of Forkhead box O1(FOXO1) and T cell clonal expansion.208 miRNAs also actively participate in B cell development. For instance, miRNA-150 dramatically impaired B cell expansion via suppressing the critical TF required for B cell differentiation, i.e., c-Myb;209 miRNA-181a promoted B cell differentiation in mouse bone marrow when ectopically expressed in B cell progenitors210 besides regulating TCR signaling in immature T cells.211 It has been documented that transplanting bone marrow cells over-expressing miRNA-181a to lethally irradiated mice promoted the growth of CD19+ B cells and reduced the amount of CD8+ T cells.210
miRNAs modulating cytokine microenvironment
Besides targeting TLR signaling pathways and regulating immune cell development, multiple lines of evidence have unambiguously supported the essential roles of miRNAs played in modulating cytokine homeostasis. It has been well documented that altered secretion profiles of cytokines such as IL2, IL6, IL10, and regulated on activation normal T cell expressed and secreted (RANTES) play crucial roles in SLE development, and miRNAs participate in SLE development through modulating the production of these primary cytokines. For example, the level of RANTES was documented to be abnormally over-represented in the blood sera of SLE patients, whereas that of IL2 was reported to be significantly lower in lupus T cells. Under-expressed miRNA-31 contributed to the decreased IL2 expression in PBMCs or lupus T cells.175,179 miRNA-142-3p was highly induced in DCs in response to LPS stimulation, leading to suppressed IL6 production.212 Up-regulated miRNA-21 expression has been positively associated with SLE activity, reduced level of which in SLE CD4+ T cells led to decreased IL10 production.178 Through characterizing miRNAs lowly expressed among SLE patients, miRNA-125a was found capable of reducing T cell-mediated RANTES production via targeting its TF, i.e., Kruppel-like factor 13 (KLF13), and exogenously introducing miRNA-125a into the T cells of SLE patients resulted in significantly alleviated up-regulation on RANTES expression and SLE severity.213
DNA methylation
DNA methylation level has been considered to be lower in SLE patients or lupus animal models.214,215 Specifically, DNA extracted from the CD4+ T cells of SLE patients was hypomethylated,215 and adoptive transfer of T cells pre-treated with DNA methylation inhibitors induced SLE symptoms in unirradiated syngeneic mice.216,217 A clinical study involving 1521 Chinese and European SLE patients, along with healthy controls and patients with other autoimmune diseases such as rheumatoid arthritis (RA) and primary Sjögren’s syndrome (pSS), revealed that SLE patients are characteristic of hypomethylation at two CpG sites within the promoter region of IFI44L, SLE patients with renal involvement displayed even lower methylation levels at these sites, and the methylation levels increased among SLE carriers during remission.218 These suggested that the methylation level of the promoter region of IFI44L may serve as the blood biomarker for SLE prognosis and diagnosis.218 In Feb 2024, the world’s first innovative IFI44L gene methylation detection product was approved by National Medical Products Administration (NMPA) of China for SLE prognosis prior to the onset of vital organ damage.219 This may be attributable to the inhibited inheritance of DNA methylation profiles during mitosis in response to perturbations such as aging and diet220,221,222 that involves the participation of multiple miRNAs such as miRNA-126, miRNA-148a and miRNA-21.179,223
DNA methylation patterns are regulated by methyltransferases, including DNA (cytosine-5)-methyltransferase 1 (DNMT1), DNMT3A, DNMT3B, and DNMT3L.224,225 While, DNMT1 maintains DNA methylation profiles, DNMT3A and DNMT3B introduce de novo DNA methylation, and DNMT3L assists the functionalities of DNMT3A and DNMT3B.226,227 It has been reported that miRNAs such as miRNA-126179 and miRNA-148a223 regulated the levels of DMNT1 in the T cells of SLE patients. Specifically, miRNA-148a and miRNA-21 were robustly up-regulated in CD4+ T cells from SLE patients lupus-prone MRL/lpr mice, giving rise to DNA hypomethylation via suppressing DNMT1 expression;223 and miRNA-126 inhibited DNA methylation in CD4+ T cells of SLE patients by binding to the 3’ untranslated region (3’ UTR) of DNMT1.179 In addition, defective ERK pathway in T cells negatively affected DNMT1 expression and enhanced the development of anti-dsDNA antibodies in transgenic mice,228 suggesting the involvement of suppressed ERK signaling in priming DNA hypomethylation among SLE carriers.
Histone modification
Histone 3 (H3) and Histone 4 (H4) hypoacetylation and site-specific histone methylation alterations were found in the CD4+ T cells from SLE patients and MRL-lpr/lpr mice splenocytes.229,230 It has been reported that the histone deacetylase inhibitor trichostatin A (TSA) can restore skewed expression of IL10, IFNγ and CD154 in lupus T cells,231 and treating MRL-lpr/lpr mice with histone deacetylase inhibitors TSA and suberoylanilide hydroxamic acid (SAHA) reduced the section of IL6, IL10, IL12, and IFNγ.232,233 These findings implicated that histone modification variation contribute to the modulation of cytokine distribution in SLE pathogenesis.
Hormonal factors predisposing SLE
Female hormones such as estrogen and prolactin contribute to the activation of the immune system and thus predispose the prevalence of SLE, leading to the extreme female predominance among SLE carriers. Specifically, estrogen functions by skewing the cytokine microenvironment to favor Th2, prolactin acts via activating the immune response. Progesterone, on the other hand, represents a protective factor of SLE by damping the immune activating signals.
Estrogen
Estrogens have been reported to potentiate Th2-mediated diseases including SLE by inhibiting the production of Th1 pro-inflammatory cytokines such as IL12, TNFα and IFN({rm{gamma }}), and stimulating the secretion of Th2 anti-inflammatory cytokines such as IL4, IL10, and TGFβ.68
Estrogens include estrone (E1), estradiol (E2), and estriol (E3), with E2 being the primary biologically active estrogen. Estrogen receptors (ERs), both nuclear and membrane bound, have been identified in various types of cells involved in the innate and adaptive immune responses.234 While the nuclear ERs include ERα and ERβ, the membrane-bound ER is the G-protein-coupled estrogen receptor (GPER).235 ERs contain three functional domains, i.e., trans-activation domain, DNA-binding domain, and ligand-binding domain.236 During nuclear ER signaling, ERα and ERβ typically act in an opposite fashion in response to E2 treatment and regulate almost distinct sets of genes, with only 38 out of 228 genes being regulated by both ERα and ERβ.237 Nuclear ER signaling can be long-term and manifest genomic information via transcriptionally regulating a plethora of factors including, e.g., cytokines such as IFNs and signaling pathways such as JAK/STAT signaling.234,238,239 Different from nuclear ER signaling, GPER-mediated signaling is rapid and nongenomic. Specifically, the signal transduction cascade is initiated via intracellular calcium and cyclic adenosine monophosphate (cAMP) induction, and leads to activated phosphoinositide 3 kinase (PI3K)/ AKT and mitogen activated protein kinase (MAPK)/ extracellular signal regulated kinase (ERK) signaling.235
Estrogen contributes to the polarization of the cytokine environment to the Th2 state, where a shift of the balance between Th1 and Th2 subsets to Th2 dominance is characteristic of SLE. Specifically, low doses of estrogen promote Th1 responses for increased cellular immunity, and high doses of estrogen elevate Th2 responses for stronger humoral immune responses.240,241 This effect of estrogens is achieved via altering the Th cytokine profile from a Th1-dominant state (IL12, IFNγ, TNFα) to a Th2-dominant profile (IL4, IL6, IL10, TGFβ).68 For instance, E2 as well as E1 and E3 have been shown to stimulate TNFα secretion at low concentrations and inhibit it at high concentrations,242 the effect of which on IL10 production was shown to be the opposite.243
Prolactin
Prolactin, with increased levels detected in the serum of SLE patients, functions as both a hormone and a cytokine. Prolactin has been shown capable of stimulating almost all primary players in the innate and adaptive immune responses such as T cells, B cells, DCs, natural killer (NK) cells, macrophages, neutrophils, and hematopoietic stem cells according to a collection of in vitro, in vivo and clinical evidences. Though prolactin disruption is not essential for the normal development and functionality of the immune system,244 prolactin can synergize with IL2 in B cell activation and differentiation.245 In addition, the prolactin receptor is expressed on human immune cells including T and B lymphocytes and monocytes,246 suggestive of its promotive roles in SLE development.
Progesterone
Pregnancy-associated changes in progesterone signaling has been considered important for innate immune surveillance and tolerogenic response, rendering progesteron a protective factor of SLE. Specifically, progesterone reduces the secretion of proinflammatory cytokines such as TNFα, IL1β and IL12, leading to attenuated activities of primary players involved in both the innate and adaptive immune responses such as macrophages, DCs, CD4+ and CD8+ T cells.239
Extrinsic factors predisposing SLE
The contribution of extrinsic risk factors to SLE susceptibility increases with age, as a greater contribution of known SLE genetic risk alleles (especially residing in non-HLA genes) were found among children who developed SLE than adult SLE patients.247,248
Extrinsic trigger of SLE can be primarily classified into three categories based on their effects on SLE pathogenesis, i.e., events introducing immune activators, perturbing cytokine microenvironment homeostasis, and inducing inflammation.8
Events introducing immunoreactants
EBV infection can increase the amount of EBV nucleic acids in the blood of SLE patients,249 which activates the innate immunity and B cell differentiation by expressing type I IFNs and stimulating the production of autoantibodies specific for EBV-encoded proteins.250,251 It is worth noting that EBV infection predisposes to SLE development but not vice versa, as the serum anti-EBV capsid antigen IgG levels of SLE patients were significantly higher than healthy individuals that did not apply to anti-EBV nuclear antigen.252 The mRNA/DNA vaccines may also induce SLE. It has been reported that the application of mRNA or DNA vaccines against the coronavirus disease (COVID-19) has been shown capable of causing new or relapsed onset of SLE.253,254,255,256 A boost in spike protein-specific CD4+ Th1 and CD8+ T cell responses were detected after the use of AZD1222 (i.e., a DNA COVID-19 vaccine),257 the mechanism of which could be attributed to activated TLRs followed by induced type I IFNs-mediated signaling. Additionally, agonists of TLR7 and/or TLR9 have been oftenly supplemented as the adjuvants in mRNA/DNA COVID-19 vaccines for enhanced immunity,258,259 further aggregating the development of SLE.
UV light irradiation may activate the autoimmune response via generating nucleic acid fragments, attributing to its breakage role on DNA strands.46 A clinical study examined the sensitivities of 100 SLE patients to UV radiation, where 93% patients showed abnormal reaction to UV and visible light including, e.g., superficial perivascular lymphocytic infiltrate and deposition of immunoreactants such as IgG and C3.260
Medications with pro-inflammatory roles may also induce SLE that can be manifested as vasodilation and hypotension. For instance, hydralazine (a vasodilator) and procainamide (an anti-arrhythmic agent) can trigger SLE via forming neutrophil extracellular trap (NET)261,262 that can function as the auto-antigens due to DNA, histones and neutrophil proteins it contains.263 Excessive secretion of pro-inflammatory cytokines such as IFNα (used for treating Hepatitis B/C) aggravates SLE.264,265,266
Habits such as tobacco smoking is a known risk factor for SLE in a dose-dependent manner as it is a stimulus capable of inducing nonspecific inflammation and, thus, autoimmune responses among SLE carriers.267
Events skewing cytokine microenvironment
Some medications may induce SLE, though the symptoms may be milder than idiopathic SLE. The mechanisms-of-action may be attributed to their roles in skewing the cytokine microenvironment. For instance, carbamazepine, an anticonvulsive agent traditionally used for treating epileps and neuropathic pain, can increase IL5 secretion that marks Th2 production, attributing to the terminal metabolite acridine it produced.268 Sulfasalazine, used for treating rheumatoid arthritis, can skew the cytokine microenvironment to Th2-dominant state by suppressing IL12 production in macrophages.269 Another example refers to antibodies against TNFα that have been used as immunosuppressors in the treatment of autoimmune or inflammatory diseases. Infliximab, an anti-TNFα antibody, induced the production of anti-dsDNA antibody and SLE among more rheumatoid arthritis patients.270,271 Similarly, hydralazine, procainamide and other DNA methyltransferase inhibitors such as 5-azacytidine may turn CD4+ T cells autoreactive to spontaneously lysed syngeneic macrophages and produce more IL4/6 and IFNγ to induce SLE.216,220 The combinatorial use of minocycline (a semi-synthetic tetracycline-class broad-spectrum antiboitic) with bone marrow derived mesenchymal stem cells (MSCs) in treating autoimmune encephalomyelitis, though having achieved desirable therapeutic effects in an autoimmune encephalomyelitic mice model, may increase the risk of developing SLE syndromes as a result of suppressed production of IFNγ and TNFα as well as increased generation of IL4 and IL10.272 Paradoxically, minocycline decreased C-C motif chemokine ligand 22 (CCL22) production from macrophage type 2 for reduced Th2 recruitment to the lesion,273 implicating the importance of cytokine microenvironment homeostasis in preventing autoimmune syndromes that is dictated, at least partially, by the type and cytokine profile of the syndrome as well as the medication strategy being applied.
Preventive strategies for SLE management
SLE could be initiated and accelerated by complicated dynamic interplays between intrinsic and extrinsic factors. Specifically, once individuals possessing SLE genetic, epigenetic or hormonal risk factors are chronically exposed to extrinsic risk factors, accelerated disease onset and deterioration may occur. As intrinsic risk factors including those at the genetic, epigenetic, hormonal levels are difficult to control, we focus on preventive approaches against the extrinsic risk factors in this section.
Extrinsic factors can be environmental situations such as virus infection, UV light irradiation, heavy metal exposure, air pollution and silica, habitual factors such as unhealthy diet, cigarette smoking, lack of physical exercises and sleep deprivation, physiological conditions such as comorbidities, obesity and pregnancy, and psychological factors such as trauma and stress.274,275,276 Current conceptions on a healthy lifestyle with a lower risk of developing SLE overall include, e.g., a healthy eating habit (i.e., top 40% of the Alternative Healthy Eating Index), no smoking, moderate alcohol consumption (i.e., no less than 5 gm per day), regular exercise (performing at least 19 metabolic equivalent hours of exercise per week), and fitness (i.e., body mass index below 25 kg/m2).277 Each of these preventive recommendations has a 19% additive value in reducing the chance of developing SLE especially among anti-dsDNA antibody positive patients; and the cumulative risk of having SLE can be reduced to half of those with the poorest behavior for individuals keeping the best adherence to the healthy lifestyle. This implicates that extrinsic factors may act synergistically to influence the risk of SLE, and SLE may be prevented by, e.g., altering the lifestyle among other extrinsic factors.
Preventive strategies can be classified into three stages, i.e., primary prevention, secondary prevention, tertiary prevention, which should be adopted as early as possible to prevent the development, exacerbation and progression of SLE, respectively. This especially holds true for individuals who have already been prognosed at a high risk of developing SLE or diagnosed with SLE.
Following the rationals of mechanisms-of-action, preventive strategies can be classified into approaches against events introducing immune stimulants, and skewing the cytokine microenvironment (Fig. 5).
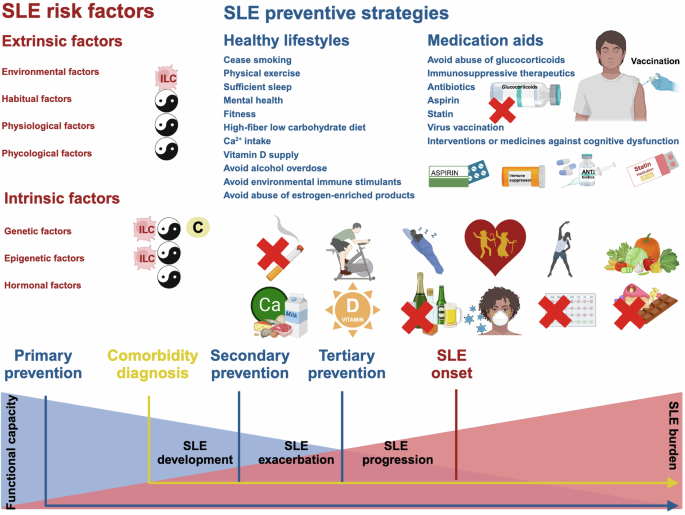
Risk factors and preventive strategies against SLE development, exacerbation, progression. Risk factors predisposing SLE can be classified into intrinsic and extrinsic factors (in red). Intrinsic factors can occur at genetic, epigenetic and hormonal levels. Extrinsic factors can be environmental, habitual, physiological and phycological. Among these extrinsic factors, SLE comorbidities from the category of physiological factors include cancers, cardiovascular diseases, bone diseases and neuropsychiatric diseases. As intrinsic SLE predisposing factors are difficult to control, preventive strategies against each extrinsic risk factors are listed (in blue). By summarizing preventive strategies against SLE and its comorbidities, 11 recommendations on the lifestyles (i.e., cease smoking, physical exercise, sufficient sleep, mental health, fitness, High-fiber low carbohydrate diet, Ca2+ intake, vitamin D supply, avoid alcohol overdose, avoid environmental immune stimulants, avoid abuse of estrogen-enriched products) and 7 advises on medication aids (avoid abuse of glucocorticoids, immunosuppressive therapeutics, antibiotics, aspirin, statin, virus vaccination, interventions of medicines against cognitive dysfunction) are given. SLE preventive strategies can be classified into three stages, i.e., primary prevention against SLE development, secondary prevention against SLE exacerbation, tertiary prevention against SLE progression. The prevention of SLE comorbidities should start at the time of diagnosis. Applying preventive strategies as early as possible during the disease course can help to avoid organ damage, which is a major trigger of systematic functional decline. Primary prevention recommendations are emphasized using cartoons
Prevention of SLE
Preventive strategies recommended below are categorized by the pathogenic stages leading to SLE. These approaches are suitable to individuals diagnosed with high risks of developing SLE for prevented disease onset, and to SLE patients for alleviated disease symptom or delayed disease progression.
It is worth to highlight that intrinsic and extrinsic factors especially biomarkers at the genetic and epigenetic levels as aforementioned can aid in SLE prevention and early intervention, since they could help identify individuals with the high risk of developing SLE that require early adoption of prevention strategies.
Also deserves emphasizing is that there is current no consensus on strategies for SLE prevention given the complexity and heterogeneity of the etiology of this disease. Besides, the full spectrum of preventive strategies against SLE is not completely uncovered, personalized prognosis and prevention modality design are not yet available. Thus, approaches beneficial to one individual may not be optimal or even effective to another.
Prevention against events introducing immune stimulants
As an important type of extrinsic factors, environmental situations with SLE-predisposing roles such as EBV infection, exposure to UV light, heavy metals such as mercury,278 agricultural pesticides,278,279,280 air pollution, crystalline silica dust,281,282,283,284 and other respiratory particulates285,286 largely act via exposing individuals with immune stimulants. These factors function primarily by stimulating cellular necrosis and secreting intracellular antigens for up-regulated IFN levels and promoted inflammation. Take EBV infection as an example, it functions by releasing EBV-encoded small RNA from infected cells that induces type I IFN signaling (in particular TLR3-mediated signals),287 with a substantially higher seroprevalence of anti-viral capsid antigens IgG and antibodies against early antigens being observed among SLE patients as compared with non-diseased controls according to a meta-analysis involving 25 case-control studies.252 Additionally, UVB radiation, another important trigger of SLE, led to a significant rise in type I IFN signaling and prolonged activation of T cells both in lupus-prone mice and among SLE patients.288,289,290 A randomized, vehicle-controlled, double-blind clinical study including 25 CLE patients revealed the photoprotective effects of broad-spectrum sunscreen.291 There also exists clinical evidence supporting the benefits of using sunscreen for preventing the onset or worsening of SLE.292 Yet it is worth noting that the association of UV radiation with SLE risk is convolved with its positive roles in vitamin D3 synthesis293 that may, to some extent, reduces SLE risk.294 Therefore, keeping away from immune stimulants such as virus infection, overt UV light exposure and any environmental pollutant is highly advised among individuals genetically predisposed with SLE.
Receiving vaccination to elevate the thresholds of responding to environmental stimuli may represent another useful strategy. Yet, the efficacy of vaccination in reducing SLE risk remains to be elucidated.295 This is because that vaccines may contain elements such as molecular mimicry, auto-antibodies and adjuvants that can potentially trigger autoimmune responses towards accelerated SLE. Evidences supporting this rational include a series of reports on the new-onset of autoimmune diseases including autoimmune hepatitis disease,296 autoimmune thrombotic events,297 rheumatoid arthritis,298 immunoglobulin A vasculitis,299 Guillain-Barré Syndrome and,300 importantly, SLE,255,301 after getting vaccinated against COVID-19. Also, a clinical case on the transition of cutaneous lupus erythematosus (CLE) to SLE after COVID-19 vaccination has been identified.302 However, these reports are largely from case reports or cross-sectional studies representing temporal associations and are not from vaccinations against EBV. On the other hand, therapeutic EBV vaccines have been considered promising in treating cancers with acceptable toxicity.303 Take together, establishing vaccines against EBV or any other immune stimulants for SLE prevention may be worthwhile to try but requires intensive investigations and clinical monitoring on the possible adverse effects accompanied besides the efficacy.
Prevention against events skewing cytokine microenvironment
Many lifestyles and physiological conditions associated with SLE predisposition can increase levels of pro-inflammatory cytokines, leading to skewed cytokine microenvironment.
Cigarette smoking has been associated with increased risk of developing anti-dsDNA antibody positive SLE than non-smokers by several clinical investigations,267,304,305 linked to augmented autoreactive B cells by a clinical evidence based meta-analysis,267 and associated with induced pulmonary ANA in the lungs of exposed mice.306 In addition, a cross-sectional study containing 105 smokers revealed the positive association of cigarette smoking with cumulative chronic damage in SLE patients and its deleterious effects on lupus morbidity.307 This may be attributed to the toxic components of the cigarette that can damage DNA to form immunogenic DNA adducts for promoted production of anti-dsDNA antibody and pro-inflammatory cytokines.308,309 In particular, smoking can increase the expression of B lymphocyte stimulator (BLyS) that is a soluble ligand of the TNF cytokine family,306 TNFα and IL6.310 Among positive ANA women, elevated BLyS and lower IL10 (an anti-inflammatory cytokine) levels were identified among frequent smokers.311 Therefore, cease smoking is highly recommend especially for those with genetic preposition to SLE.
Sleep-deprivation (less than 7 h per night) has also been associated with an earlier SLE onset and accelerated auto-antibody production from both pre-clinical animal models and clinical observations.312,313,314 In particular, from a prospective study involving 436 participants having relatives already diagnosed with SLE, the chance of developing SLE among individuals sleeping less than seven hours per day was 2.8-folds of that among the rest with statistical significance.314 In a larger trial involving 186072 women, chronic low sleep duration (i.e., less than 5 h/day) was associated with increased SLE risk (adjusted HR = 2.47) with confounding factors shiftwork, bodily pain and depression being adjusted.315 At the molecular level, insufficient sleep has been shown to be capable of increasing levels of pro-inflammatory IL6 and TNFα, leading to impaired functionalities of Treg cells, imbalanced cytokine milieu, systematic inflammation and ultimately endangered self-immune tolerance.278 Thus, keeping sufficient sleep (e.g., seven to eight hours per day) and avoiding night or rotating shifts are recommended for individuals of all ages predisposed with SLE.
Adipose tissue especially visceral fat can secrete pro-inflammatory cytokines such as IL6 and possess high levels of TNFα receptor 2 (TNFR2) as compared with non-obese individuals.316 In addition, both TNFα and IL6 have been implicated with critical roles in the modulation of insulin resistance.317 In a Mendelian randomization study, 3 out of 25 identified cytokines associated with obesity (i.e., CTACK, IL18 and SCGFb) were related to SLE, and 2 out of 18 characterized cytokines associated with SLE (i.e., IP10 and MIP1B) were also linked to obesity,318 implicating the intermediary role of these cytokines in the association between obesity and SLE. Thus, keeping fitness may be an effective preventive approach against SLE. To avoid obesity, one might be recommended to take food enriched with fibers and/or adhere to a low-carbohydrate or ketogenic diet that have been shown associated with reduced risk of developing SLE via decreasing glucose-induced inflammation and preventing obesity.319,320
Women who had experienced physical and emotional abuse during childhood exhibit higher chances of developing SLE as compared with those who had not.321,322 In an analysis of data from 36152 black women, both physical and sexual abuse during childhood were statistically associated with increased SLE incidence, where other factors including alcohol consumption, smoking, body mass index, oral contraceptive use, age at menarche, and parental education were adjusted. The HR of women with more than 2 episodes of sexual abuse was 2.51-folds of the control group, implicating the significant promoting role of physical and emotional abuse during childhood on SLE risk.321 In consistent with this, psychosocial trauma and associated post-traumatic stress disorder may lead to autoimmune diseases among women including SLE.323 In a cross-sectional analysis of data from the California Lupus Epidemiology Study involving 242 SLE adult patients, more recent stressful events were statistically associated with higher stress among people having trauma and adverse childhood experiences, and positive psychosocial factors showed an opposite association.324 These results suggested the mitigating role of a positive mental state in perceived stress that may improve outcomes in SLE, even among individuals suffered from prior trauma and thus more vulnerable to recent stressful events. Besides, depression has been recognized as an important risk factor of SLE among females.325 In a cross-sectional study conducted among 85 Chinese SLE patients, a robust correlation was identified between SLE disease activity and depression that was weakened during disease remission; besides, the presence of mucosal ulcer significantly increased the risk of developing depression among these patients.326 These results supported the negative role of depression on SLE risk. Mechanically, Th2 cytokines are generated to protect the organism from over-production of the pro-inflammatory Th1 cytokines in response to stress; under certain circumstances, stress hormones may promote inflammation via inducing the production of cytokines such as IL6 and TNFα as well as through activating pathways involving corticotropin-releasing hormone and substance P induced histamine.327 The stress system, being either hyperactive or hypoactive, may contribute to SLE pathogenesis, the process of which involves an abnormal interface between the neuroendocrine and the immune systems.327 Thus, conditions associated with dramatic changes in the stress system such as acute or chronic stress, pregnancy and the postpartum period may potentiate SLE via altering the Th1/Th2 cytokine homeostasis.327 These have collectively explained the predisposing roles of emotional depression and stress on SLE onset, and suggested the importance of keeping the mental health among individuals at high risks of developing SLE.
Estrogen has been considered capable of up-regulating the expression of several genes involved in B cell activation and survival such as those encoding CD22, Src homology 2 (SH2) domain-containing phosphatase 1 (SHP1), B-cell lymphoma 2 (BCL2), and vascular cell adhesion molecule (VCAM),328 explaining the increased SLE susceptibilities of oral contraceptive pill (with ethinyl estradiol being the major ingredient) users and individuals receiving hormone replacement therapy.329,330,331 In a two-sample Mendelian randomization analysis of a GWAS data, a negative causal relationship was identified between age at menarche and SLE, suggesting the positive role of estrogen played in SLE risk predisposition.332 Thus, reducing the use of estrogen-enriched products is highly recommended for women with genetic SLE predisposition.
Prevention of SLE comorbidities
SLE comorbidities should be prevented or treated, which is also of the paramount importance as these conditions can lead to the ultimate death in a considerable proportion of SLE patients.333 Diseases typically accompanied with SLE include neoplasms especially hematological cancers, cardiovascular diseases, bone diseases such as osteonecrosis, and neuropsychiatric manifestations such as cognitive dysfunction.
Cancers
SLE-associated cancers can mostly be induced by virus infection. The International Agency for Research on Cancer (IARC) has implicated virus infection as one primary etiological factor leading to cancers that contributes to around 15% of new cancer annual incidence. Viruses capable of inducing cancers are defined as Group I ‘carcinogenic to humans’ by IARC, which include, e.g., human papillomavirus (HPV), hepatitis B virus (HBV), hepatitis C virus (HCV), EBV, Kaposi’s sarcoma herpesvirus (KSHV), Merkel cell polyomavirus (MCPyV), human immunodeficiency virus (HIV), human T-cell lymphotropic virus 1 (HTLV1), simian virus 40 (SV40).334,335,336,337 In particular, approximately 10% diagnosed cancer cases are induced by virus infection, among which 4.5% are caused by HPV infections.338
Among HPV-associated transformed conditions, high risk was identified for epithelial carcinoma in situ of the uterine cervix, vaginal cancer, anal cancer, non-melanoma skin cancer, bladder cancer, liver cancer, lung cancer, and lymphoma especially Hodgkin’s lymphoma.339,340,341,342,343 The association between HPV infection and cancer incidence has been proposed to be attributable to the chronic immune activation triggered by persistent virus infection and/or the inefficiency of the immune system to clear off HPV infection.341 Therefore, frequent HPV screening, vaccination following gender- and age- associated recommendations, therapeutics removing immune stimulants leading to over-activated immune responses, and medications attenuating chronic over-activated immune system are all helpful for preventing the occurrence and deterioration of SLE-associated cancers.
Specifically, regular gynecological screening, such as the Papanicolaou (PAP) test for women aged 21-30 years old every 3 years and the PAP test combined with the HPV test for women aged 30-65 years old every 5 years, are heavily recommended. Along with the examination, HPV vaccination is suggested among females aged between 21 and 65.339
The traditional strategy used to remove immune stimulants such as bacterial infections is to apply antibiotics. For instance, trimethoprim-sulfamethoxazole(TMP-SMX) is a combinatorial antibiotic used for infection treatment, which has been used as a prophylactic agent for patients showing a low T cell activity (i.e., CD4+ cells below 200 cells/mm3).344 However, co-administration of TMP and SMX is not recommended due to its notable adverse effects especially for the increased risk for hematologic toxicity.345 In addition, treating patients carrying chronic neutropenia (below 500 cells/mm3) with quinolone antibiotic such as levofloxacin (500 mg daily) or ciprofloxacin (500 mg, twice daily), possibly coupled with anti-fungal therapy as well depending on the disease situation, has been recommended as another prophylactic therapy.346,347
Medications used for attenuating chronic over-stimulated immune response is to apply immunosuppressive therapies. Agents fell into this category can be sub-classified into three groups based on their effectiveness and toxicity. That is, agents taking actions rapidly but having severe long-term side effects such as glucocorticoids, agents functioning slowly but showing high safety such as hydroxychloroquine, and agents having high non-response rates and unavoidable adverse effects such as cyclosporine A, cyclophosphamide, mycophenolate mofetil, azathioprine, methotrexate, tacrolimus, belimumab and rituximab.348 Specifically, glucocorticoids, steroid hormones produced from the cortex of adrenal glands, dampen the innate immune response via attenuating signaling mediated by pattern recognition receptors (PPRs) and resolving the inflammatory response, as well as regulate adaptive immunity through inhibiting T cell activation and B cell production;349 hydroxychloroquine attenuates T cell activation via suppressing TLR signaling, inhibiting cytokine production, and reducing the expression of CD154 (a marker of antigen-specific activation of CD4+ T cells350);351 cyclosporine A, a cyclic undecapeptide commonly used as a member of third group, inhibits T cell activation via blocking the synthesis of ILs including IL2.352
Cardiovascular diseases
Cardiovascular disorders represent another important portfolio of SLE comorbidities. It has been estimated that SLE patients have a 27% higher risk of developing cardiovascular diseases as compared with gender- and age- matched diabetes patients and over double folds the risk of the general population.353 Among the diversified types of cardiovascular events, myocardial infarction and stroke are the most commonly observed among SLE carriers, with the hazard ratio of myocardial infarction among SLE patients ranging from 2.6 to 5.1 and that of stroke SLE carriers being 2.1 to 3.3.354 In addition to the aforementioned two primary cardiovascular disorders, the chance of developing sub-clinical atherosclerosis also increases among SLE patients, with the atherosclerotic plaques being detected in 25–56% young SLE carriers as compared with 17–70% of individuals from the general cohort with a similar age range.355,356,357
Several SLE-associated with cardiovascular comorbidities have been identified. These include, e.g., SLE-specific genetic risk factors such as the IL19 risk allele associated with a 2.3-fold increased risk of developing myocardial infarction and stroke among SLE carriers due to enhanced levels of IL10 and aPL antibodies,358 physiological risk factors such as male gender,359 advanced age,359,360,361 and postmenopausal status,360,361 lifestyle risk factors such as tobacco smoking,362 and medical risk factors such as hypertension,360 hypercholesterolemia, diabetes mellitus, and the associated medical interventions.
As genetic and physiological risk factors are difficult to control, preventive strategies against SLE-associated cardiovascular comorbidities largely focus on removing risk factors from the perspectives of the lifestyle and medication. Current recommendations for managing cardiovascular diseases associated with SLE are, in general, similar to the general strategies used for cardiovascular disorder management. These include, e.g., ceasing smoking and taking physical exercises for a healthy lifestyle, and relying on therapeutics for appropriate control of hypertension, hypercholesterolemia, and diabetes.363,364 Since it is easy to understand the benefits of forming healthy habits, we focus on the medicinal use for reducing the risk of developing SLE-associated cardiovascular disorders and/or halting their deterioration.
Several medications have been identified to be feasible for preventing cardiovascular diseases associated with SLE. For instance, an appropriate use of statin has been proposed capable of helping prevent cardiovascular diseases among SLE patients given its roles in reducing the low-density lipoprotein levels of the patients.363 There also exists evidence supporting a dose-dependent use of aspirin among SLE carriers especially those having at least one risk factor predisposing the onset of cardiovascular diseases due to its anti-thrombotic effects.365 Specifically, the anti-thrombotic effects of aspirin are related to its roles in acetylating cyclooxygenase (COX) in the platelets,366 and a low-dose aspirin regimen (i.e., over 30 mg/day) can effectively prevent platelet aggregation without affecting the functionalities of endothelial cells.367 Yet, a higher dosage and more frequent use of aspirin is required to suppress inflammation, the primary drawback of which is associated with increased risk of developing gastrointestinal syndromes, renal toxicity and hypertension. Taken together, aspirin is recommended as a secondary prevention approach against cardiovascular syndromes associated with SLE but remains controversial to be used for primary cardiovascular disease prevention due to its adverse effects.368
It is important to note that several drugs commonly used for treating SLE or certain comorbidities may induce or deteriorate cardiovascular syndromes. For instance, glucocorticoids, capable of dampening both the innate and adaptive immune responses, are known to increase the glucose and cholesterol levels of the blood as well as the blood pressure, which are all risk factors predisposing the onset of cardiovascular disorders.369,370 Specifically, several cohort investigations have demonstrated that a long duration use of corticosteroid371,372 and a high accumulated corticosteroid dose,372,373,374,375,376,377,378,379 are associated with a high incidence of cardiovascular events among SLE carriers.362,380,381,382
Bone diseases
Bone diseases especially osteoporosis and fractures frequently occur among SLE carriers, with the frequency ranging from 3 to 40%.120,383,384,385 In particular, independent cohort studies conducted in Taiwan, Sourth Korea, and USA all reported higher incidences of osteoporotic fractures among SLE patients as compared those non-SLE carriers, with the incidence ratio being 1.63 versus 0.92,386 19 versus 6.5,387 and 4.32 versus 2.4,388 respectively, per 1000 persons per year.
Several risk factors have been alluded to increase the chance of developing osteonecrosis among SLE patients, with the most prominent one being the medicinal use of corticosteroids. It has been recognized that osteonecrosis primarily occurs among SLE patients who have received corticosteroid treatment but not those who have not.389 Besides, the prevalence of osteonecrosis among SLE is much higher than in other diseases requiring the use of corticosteroids.390 Lots of clinical evidence have implicated that the initial dose, cumulative dose, tapering speed, and treatment duration of corticosteroids, as well as medications used together with corticosteroids all convey significant impact to the incidence, number, volume and location of osteonecrosis.390 Thus, avoiding the use or overt dosage of corticosteroid represents an effective strategy for keeping bone health.
Habits such as alcohol abuse and smoking all impose significant negative impacts on the onset of SLE-associated osteonecrosis.391,392,393 Alcohol intake is associated with induced chronic inflammation, impaired bone cell differentiation, and increased adipogenesis, which collectively lead to the development of necrosis including osteonecrosis.394 Smoking is capable of inducing osteoblast apoptosis that leads to decreased bone mass.395 As preventive strategies, SLE patients are suggested to reduce the amount of alcohol intake and cease smoking, increase calcium (Ca2+) and vitamin D intake, and increase physical activities to prevent the onset and deterioration of SLE-associated osteonecrosis. For patients at the risk of fractures, medications against bone resorption or destruction may be considered such as bisphosphonates and denosumab.396
Lastly, accumulated evidence have implicated that SLE activity is of the paramount importance in predisposing osteonecrosis.397,398,399,400,401 Several clinical studies have supported the positive correlation between the SLEDAI score and the chance of developing osteonecrosis.398,402 Several genetic polymorphisms affecting SLE activities have been identified to be positively associated with the risk of developing osteonecrosis. For example, three SNPs from the Korean population (i.e., rs3813946, rs311306, rs17615)403 and one SNP from the Chinese population (i.e., rs45573035)404 residing in the gene encoding the complement receptor type 2 (CR2) have been reported to be associated with the susceptibility of osteonecrosis. CR2 is a membrane glycoprotein that binds to the degradation debris of C3 and plays critical roles in relaying immune signals such as activating B cells via cooperating with the B cell receptors.403,405 Genetic defects in CR2 may lead to impaired debris clearance ability and abnormal B cell signaling, which are both promotive on SLE activity. Another example is that the T/T genotypes from the C1236T and C3435T polymorphisms of the gene encoding the adenosine triphosphate (ATP)-binding cassette subfamily B member 1 (ABCB1) show protective roles against osteonecrosis as compared with the wildtype (C/C) genotypes, where ABCB1 functions by pumping foreign substances out of cells and is lowly expressed among SLE patients.406,407,408 In summary, reduced level of ABCB1 is associated with cells’ decreased ability of removing immune stimulants and consequently increased SLE activity and elevated risk of developing osteonecrosis. Thus, adopting preventive strategies against SLE is highly recommended here for maintaining the bone health.
Neuropsychiatric diseases
Neuropsychiatric disorders, ranging from overt neurological syndromes such as psychosis and seizures to subclinical conditions such as mood disorders and cognitive dysfunction (defined as a significant defect in attention, memory, language, reasoning, execution, visual-spatial processing, and psychomotor speed409), represent an important type of SLE comorbidities. Among these varied disease manifestations, cognitive dysfunction has the highest prevalence, with up to 50% SLE patients carrying overt neurological syndromes being accompanied with varied degrees of cognitive dysfunctions.410 The overall prevalence of cognitive dysfunction varies between 3% and 88% among SLE patients411,412,413,414 due to the lack of consensus in screening tools and validated markers for identifying cognitive dysfunctions, difficulty in associating cognitive dysfunctions with SLE, and heterogeneity regarding the severity and cohort intrinsic feature of SLE patients.410,415,416,417,418,419 Clinically, only 3-5% SLE patients carry severe cognitive dysfunctions, and the degrees of the cognitive syndromes of most patients are mild-to-moderate following a benign course.420
Conventional neuroimaging have shown that, although varying significantly among individuals, cognitive dysfunctions primarily display periventricular hyperintensities and cerebral atrophy.421 The pathogenesis of SLE-related cognitive dysfunctions are considered to be caused by the access of peripherally produced neurotoxic auto-antibodies to the central nervous system, the process of which involves the breach of the blood-brain barrier (BBB).422 Therefore, neuropsychiatric disorders can be roughly considered as a manifestation of cardiovascular syndromes commonly occurred among SLE carriers in the brain system, leaving risk factors predisposing the development of neuropsychiatric and cardiovascular diseases associated with SLE similar. Besides physiological factors that are difficult to manage by nurture, the negative impacts of some lifestyles and medical predisposing factors of cardiovascular comorbidities should be removed to keep the mental health of SLE carriers. These include the avoidance of smoking423 and abused use of glucocorticoids as aforementioned. Of note is that both long- and short- term application of glucocorticoids for SLE control have been shown capable of worsening cognitive dysfunction among SLE patients by several clinical studies.424 For instance, long-term use of moderate doses of prednisolone led to reduced cognitive flexibility and decision-making ability of SLE patients;425 a daily use of prednisolone for over 9 mg worsened the mathematical processing ability of SLE patients with SLE;426 short-term application of high-dose glucocorticoid resulted in hippocampal atrophy and declarative memory defects.427 The mechanistic impact of glucocorticoids on the cognitive system of SLE patients can be attributed to the interactions of these agents with the neuronal receptors distributed in the prefrontal cortex, hippocampus and basolateral amygdala, which lead to impaired memory and learning activities.428,429 On the other hand, several observational studies have supported the utility of aspirin in rescuing cognitive dysfunctions.430,431,432,433,434,435 For instance, regular administration of low-dose aspirin improved the cognitive function of the 123 SLE patients recruited from a 3-year prospective study, with a particular beneficial impact observed among the elder individuals also possessing cardiovascular risk factors such as diabetes.430 However, no consensus has been reached regarding the use of statin436 for treating cognitive dysfunctions according to current clinical settings as has been recommended for preventing cardiovascular comorbidities.
In addition, individualized multipronged strategies specifically designed for mental health management have been recommended for preventing cognitive dysfunctions. These can be categorized into non-pharmacological and pharmacological approaches.
Non-pharmacological interventions include the engagement of regular physical activities and cognitive rehabilitation.422 Regular physical activities encompass three lifestyle components, i.e., mental, physical, social,437,438,439 which regain the cognitive functionalities via, e.g., improving neurovascular coupling that is defined as increased neuronal activities linked to the local regulation of cerebral blood flow.440 The benefits of physical activities to cognitive behavior improvement is dose-dependent. That is, the risk of developing cognitive deterioration can be ameliorated by taking moderate-intensity aerobic exercises for 30 min and 5 days per week or, alternatively, by taking high-intensity aerobic exercises for above 20 min and 3 days per week.441 Cognitive rehabilitation is performed by occupational therapists to intensively retrain the cognitive and memory skills of the patients,442,443 the process of which is comprised of, but not limited to, cognitive behavior intervention, cognitive training exercises, prioritization, time optimization and memory aids.444 Cognitive behavior intervention aims at helping SLE patients with self-perceived cognitive dysfunctions to improve their memories and the abilities to perform daily activities.445 Cognitive training exercises such as chess can help the patients enhance their executive functions and problem-resolving skills.444 Prioritization can help SLE patients carrying cognitive dysfunctions to focus on one task prior to proceeding to the next. Time optimization helps the patients to prioritize cognitively intensive tasks to the earlier time of a day. Memory aids aim to help the patients to better manage their daily activities that can be in the form of, e.g., written reminders.422
Pharmacological medications used for improving cognitive dysfunctions include, primarily, N-methyl-D-aspartate receptor (NMDAR) antagonists, acetylcholinesterase inhibitors, and C5a receptor blocking agents.
The NMDAR is an ionotropic glutamate receptor mediating excitatory neurotransmission in the mammalian brain, which are localized in the postsynaptic terminal allowing for the influx of sodium (Na+) and Ca2+. NMDAR plays essential roles during synaptic transmission for synaptic plasticity and normal neuronal activities. Thus, NMDAR antagonists such as memantine can help the patients regain memory and learning abilities, and thus have been considered for managing patients carrying moderate-to-severe dementia.446
Acetylcholinesterase is an enzyme residing largely in the synaptic cleft that rapidly breaks down acetylcholine molecules to prevent prolonged action and allow for their in time recycling. Acetylcholinesterase inhibitors such as donepezil, rivastigmine, galantamine thus can help improve the cognitive abilities of the patients by increasing the availability of acetylcholines at the synaptic clefts and avoiding their overt activities.447 According to a meta-analysis involving 10 randomized, double-blind, placebo-controlled clinical trials of a 6-month treatment of acetylcholinesterase inhibitors, these agents were found to be associated with reduced cognition decline.448,449
The BBB is an unique structure in the brain for the maintenance of brain homeostasis. The complement activation byproduct C5a can elevate the permeability of BBB by relaying signals through its G-protein coupled receptor C5aR1. Thus, C5a receptor blockage therapeutics can prevent SLE patients from developing cognitive syndromes by keeping the integrity of BBB.450 It has been shown using MRL/lpr lupus prone mice that C5a receptor blockade can effectively ameliorate BBB disruption for attenuated cognitive abnormalities.451
Current therapeutics for SLE treatment
Current therapeutics attenuating immune response
TLR signaling plays a central role in the development of SLE. Endosomal TLRs (including TLR3, TLR7, TLR8, TLR9, and TLR13) that are specialized to sense nucleic acids detect self-derived damage-associated molecular patterns (DAMPs) together with surface TLRs to collectively trigger the downstream production of pro-inflammatory cytokines. Therefore, disturbing TLR signaling remains a strategy to fight against SLE. Enpatoran, being a highly selective dual TLR7 and TLR8 inhibitor, has been developed for treating autoimmune disorders including SLE, CLE and myositis. Several clinical trials including the ethno-bridging phase I trial (NCT04880213) and the ongoing phase II trials (NCT05162586, NCT05540327) have been or being performed to examine the treatment efficacy of enpatoran, as well as its pharmacokinetic, pharmacodynamic, and clinical safety.452 Current clinical and pre-clinical evidence have implicated a potential glucocorticoid-sparing effect of enpatoran for SLE treatment and the promoted overall health among SLE patients receiving enpatoran.452,453 E6742, another selective dual TLR7 and TLR8 inhibitor was found capable of ameliorating the pathogenesis features of lupus mice,454 yet its clinical evidence from a phase I/II clinical trial (NCT05278663), though been completed, has not been published. On the other hand, anti-malarial agents such as hydroxychloroquine, chloroquine, quinacrine and artemisinin can impede TLR from recognizing nucleic acids via masking TLR-binding epitopes of nucleic acids and thus be feasible for SLE treatment as well.455,456,457,458,459,460,461,462 While hydroxychloroquine has been recommended for all SLE patients due to its comparatively lower incidence of developing side effects such as retinopathy and cardiomyopathy than other antimalarial agents via inhibiting TLR3/7/9,348,463 chloroquine resolved skin lesions among CLE patients by functioning as a TLR7/8/9 antagonist,459 quinacrine ameliorated skin conditions of CLE patients through targeting TLR7/9,462 and artemisinin prevented the recurrence of LN via inhibiting TLR4 (Table 2).460
Strategies targeting type I IFNs for attenuated autoimmune response and SLE severity have been established for SLE treatment. For instance, litifilimab, an antibody against blood DC antigen 2 (BDCA2, also named CD303), which is expressed exclusively on plasmacytoid DCs464 and a major predictor of type I IFNs production,465 decreased the expression of genes relaying IFN signals among SLE patients in a phase I clinical study (NCT02106897)466 and attenuated the activities of SLE and CLE in a phase II clinical trial (NCT02847598).467,468 Anifrolumab, a human monoclonal antibody against type I IFNs receptor subunit 1 exhibited improved complete renal response among LN patients and reduced glucocorticoid use in a phase II randomized trial (NCT02547922),469 elevated the response rate from a phase III trial (NCT02446899),103 and long-term safety and tolerability in a randomized placebo-controlled phase III extension trial (NCT02794285).470 Combining the standard therapy with anifrolumab showed higher remission rates than using the standard therapy alone in SLE management according to a 4-year study.471 Attributing to these clinical successes, anifrolumab received its first approval as an add-on therapy for treating moderate-to-severe SLE in the United States in July 2021,472 in Japan in September 2021, in the Europe in February 2022, in Hong Kong in December 2022, and in the Guangdong province of China in October 2024.473,474 Medications directly targeting IFNα have also demonstrated positive results on SLE management. For instance, IFNα kinoid, a vaccine inducing neutralizing antibodies against IFNα, significantly reduced the IFN gene signature and attenuated symptoms with an acceptable safety profile in a phase IIb study (NCT02665364)108 and phase I/II trial (NCT01058343) among SLE patients.475 Anti-IFNα antibodies such as rontalizumab and sifalimumab showed reduced flares, decreased steroid use and improvement management of SLE patients in a phase II (NCT00962832) and phase IIb (NCT01283139) clinical trial, respectively476,477 (Table 2).
Strategies targeting the JAK/STAT axis have also been shown feasible for treating SLE via intervening type I IFN mediated signaling. These include, e.g., JAK inhibitors such as upadacitinib and filgotinib, as well as STAT inhibitors such as fludarabine and artesunate. Specifically, upadacitinib reduced the flares of SLE patients with good tolerance in a phase II clinical trial (NCT03978520);110 and filgotinib showed desirable therapeutic efficacy in treating lupus membranous nephropathy according to a phase II clinical trial (NCT03285711)478 and in treating moderate-to-severe CLE based on results from another phase II study (NCT03134222).479 Combined use of low-dose fludarabine and cyclophosphamide for a short duration led to long-lasting disease remission among LN patients but bone marrow toxicity from a phase I/II study;480,481 and artesunate demonstrated its therapeutic efficacy and safety in treating active lupus nephritis in a phase IV trial (NCT03214731) when being coupled with the standard of care482 (Table 2).
Strategies impairing the presentation of antigens to T cells by innate lymphocyte cells (ILCs) have also been established for SLE treatment. For example, rigerimod (a 21-mer linear peptide derived from the small nuclear U1RNP-70K) inhibits B cell maturation by reducing the stability of MHC II molecules towards blocked antigen presentation to auto-reactive T cells.483,484 Rigerimod improved the clinical symptoms of 20 moderate SLE patients,485 and slightly outweighed placebo in a phase III trial examining its clinical efficacy in treating SLE (NCT02504645).486
Immunosuppressors such as mycophenolate mofetil, azathioprine, cyclophosphamide, mizoribine, and leflunomide have been used for SLE treatment, attributing to their roles in imposing cytotoxic effects on rapidly growing cells including proliferating T and B lymphocytes.487,488,489,490 Specifically, mycophenolate mofetil obstructs the formation of guanine nucleotides via inhibiting inosine monophosphate dehydrogenase (IMPDH) and thus disturbs the generation of DNA necessary for cell replication;489 azathioprine is a purine antagonist blocking nucleotide synthesis and inhibiting leukocyte proliferation;488 and cyclophosphamide functions as an alkylating agent disrupting DNA replication.487 Mycophenolate mofetil, azathioprine and cyclophosphamide have all been widely used in the clinics for SLE management with considerable good therapeutic responses received. Yet, cyclophosphamide is more inclined to be used as an induction therapy (often applied as the first-phase therapeutics) for treating severe conditions, azathioprine is prone to maintain the disease situations, and mycophenolate mofetil serves both therapeutic purposes481,491,492,493,494,495,496,497,498,499,500,501 with comparable efficacy and reduced toxicity.500 Mizoribine, functioning similarly with mycophenolate mofetil, enhanced the clinical and serological indexes of 5 SLE patients.502 Leflunomide works as a dihydroorotate dehydrogenase (DHOH) inhibitor to interfere with pyrimidine synthesis.503 It has been shown that low-dose leflunomide effectively improved the treatment efficacy and safety of Chinese LE patients receiving prednisone therapeutics,494 and a trial involving 17 LE patients demonstrated the safety and efficacy of leflunomide in treating refractory LE patients or those having developed resistance to conventional therapies.504 Leflunomide displayed a similar efficacy with azathioprine in treating LN (NCT01172002).492 In addition, calcineurin inhibitors such as cyclosporine A, tacrolimus and voclosporin have been used for SLE management via executing their immunosuppressive functions.505,506 For instance, cyclosporine A improved the therapeutic outcome of LN receiving prednisolone (a type of corticosteroids);493 and was shown as effective as cyclophosphamide in treating LN patients according to a phase II trial (NCT00976300).496 Tacrolimus suppressed T cell activation and B cell differentiation by inhibiting the calcineurin pathway as a result of inhibited dephosphorylation and translocation of NFAT.507,508 A prospective clinical case study involving 19 patients demonstrated the feasibility and safety of long-term and low-dose use of tacrolimus for treating young LN patients;509 and a phase IV clinical trial composed of 150 LN patients and an unknown phase study reported comparable treatment efficacies between tacrolimus and mycophenolate mofetil in the long run (NCT00371319).510,511 Voclosporin, another calcineurin inhibitor,512,513 showed superior safety, efficacy and feasibility for its long-term use in phase III clinical trials against LN (NCT03021499, NCT01580865, NCT03597464).511,514,515 A phase II clinical trial demonstrated that low-dose voclosporin could serve as the induction therapy for treating active LN for enhanced renal response rate (NCT02141672) (Table 2).516
The pair of CD40 receptor and its ligand CD40L (i.e., CD40-CD40L) is one of the most critical molecular axes mediating the crosstalk between T and B cells.517 While B cells constitutively express high levels of CD40, T cells express high levels of CD40L upon activation.517 Due to the essential role of the CD40-CD40L interaction played during the adaptive immune response, agents targeting CD40 or CD40L have been established that had held a great promise. For instance, dapirolizumab pegol has demonstrated its efficacy and safety in treating SLE via targeting CD40L according to a phase II trial (NCT02804763),518,519 and is currently undergoing two phase III trials as a therapeutic against SLE (NCT04976322, NCT04294667) (Table 2).
BLyS and a proliferation-inducing ligand (APRIL) are vital survival factors regulating B cell survival, proliferation and differentiation.520 Thus, BLyS inhibitors may execute immunosuppression capacity via dampening the activation and proliferation of B cells.521 Belimumab, an inhibitor of BLyS having been approved by FDA for SLE management in 2011, has been effective in dealing with and preventing renal damages among SLE patients.522,523,524 Blisibimod, another BLyS inhibitor, has been associated with reduced steroids, decreased proteinuria among SLE patients in a phase III study (NCT01395745);525 blisibimod also significantly reduced levels of proteinuria, anti-dsDNA antibody, B cells but increased the amount of complements C3 and C4 in a phase II study (NCT01162681).526 Tabalumab, through suppressing BLyS, displayed positive therapeutic changes regarding the levels of anti-dsDNA antibody, complements, B cells and immunoglobulins according to two phase III studies (NCT01196091, NCT01205438).527,528 Several clinical trials are being conducted to investigate the efficacy of ianalumab in treating SLE as an inhibitor of the BLyS receptor.529 Atacicept, a recombinant fusion protein concomitantly blocking BLyS and APRIL, showed therapeutic fitness in treating SLE patients especially for those with high disease activity in a phase II study (NCT01972568).530 Atacicept was also associated with reduced levels of total IgG and anti-dsDNA antibody as well as increased amount of C3 and C4 in a phase II/III study (NCT00624338).531,532 Telitacicept is a fusion protein comprised of a recombinant transmembrane activator and calcium modulator and cyclophilin ligand interactor (TACI) receptor fused to the fragment crystallizable (Fc) domain of human IgG. By binding to and neutralizing the activity of BLyS and APRIL, telitacicept suppresses the development and survival of PCs and mature B cells. Telitacicept received its first approval in China for treating active SLE patients in 2021.533 Rozibafusp alfa (AMG 570) is a first-in-class bispecific IgG2-peptide fusion protein designed to target both BLyS and inducible T-cell costimulator ligand (ICOSL), where ICOSL is mainly expressed on B cells and APCs. By suppressing BLyS, rozibafusp alfa dampens the differentiation and survival of B cells and PCs. In addition, rozibafusp alfa blocks the interactions between Th cells (especially Tfh) and B cells for impaired antibody production as a result of inhibited ICOSL. The therapeutic efficacy of rozibafusp alfa was promising according to the results obtained from mouse SLE models.534 Phase I and II clinical trials examining the feasibility of using rozibafusp alfa for SLE treatment have been launched with the results not available yet (NCT02618967, NCT04058028).535 (Table 2).
Bruton’s tyrosine kinase (BTK), an intracellular signaling molecule of B and myeloid cell pathways, is of the vital importance for B cell development and activities. Activated BTK signaling is associated with elevated production of auto-antibodies that can form ICs with auto-antigens and deposit in tissues to induce inflammation and damage the lesions.536,537 BTK inhibitors such as elsubrutinib, branebrutinib, fenebrutinib and evobrutinib, thus, represent a portfolio of promising immunosuppressors coping with SLE. Elsubrutinib, in combination with upadacitinib (a JAK1 inhibitor), improved SLE clinical syndromes without obvious adverse concerns in a phase II study (NCT03978520).110 Branebrutinib, another BTK inhibitor with demonstrated in vivo evidence showing its superiority in treating SLE,538 is currently under clinical investigation (NCT04186871).539 The BTK inhibitor fenebrutinib reduced the levels of anti-dsDNA antibody and total IgG, and increased that of complement C4 among SLE patients according to a phase II study (NCT02908100).540 Evobrutinib displayed a positive long-term efficacy in treating SLE patients carrying relapsed multiple sclerosis in a phase II trial (NCT02975336).541 More BTK inhibitors are under clinical investigations regarding their fitness in treating that, include, e.g., orelabrutinib (NCT04305197, NCT05688696),542,543 zanubrutinib (NCT04643470),544 and AC0058TA (NCT03878303) (Table 2).545
Besides inhibiting B cell activation, B cell depletion represents another portfolio of strategies attenuating over-activated adaptive immune response for SLE control. CD19 and CD20 are B cell linkage-specific antigens expressed on the surface of most B cell lymphocytes, with CD19 being expressed across the entire spectrum during B cell maturation and CD20 being present during the late stages of B cell lymphogenesis.546,547 Obexelimab is a monoclonal antibody binding to CD19 and FcyRIIb, and has been reported by a double-blind, randomized, placebo-controlled phase II study capable of reducing the amount of B cells in SLE patients for improved therapeutic response and efficacy (NCT02725515).548 Therapeutics utilizing chimeric antigen receptor (CAR)-T cells to target CD19 and/or B cell maturation antigen (BCMA) expressed on B cells or their end-stage PCs have proven to be a revolutionary successful regimen for treating diseases associated with over-activated B cells including SLE.549 Mechanically, hyperactive B cells can cause inflammation and tissue damage by producing overt autoantibodies, and carefully designed CAR-T therapeutics can reset the immune system to alleviate the disease symptoms.550,551 For example, CAR-T cells targeting CD19 hold a significant promise in treating refractory SLE patients, with rapid and profound depletion of B cells and concomitant improvement in clinical symptoms and serological markers being reported.551,552,553,554,555 A phase I study employing a dual targeting strategy against CD19 and BCMA depleted both B cells and PCs, with the majority of the SLE patients showing negative results for all autoantibodies including those secreted by long-lived PCs after the treatment.555 More clinical trials on CAR-T cells targeting other B cell specific antigens or antigen combinations such as BCMA CAR-T cells (NCT06038474),556 CD20/BCMA CAR-T cells (NCT06249438),557 and CD19/CD20 CAR-T cells (NCT06462144, NCT06153095, NCT06567080) in treating SLE have been registered.558,559,560 Despite these encouraging clinical results regarding the safety and efficacy of CAR-T cell therapeutics in treating SLE patients, challenges exist that have substantially hindered the wide adoption of this promising approach for SLE management. First, the long-term persistence and functionality of CAR-based immune cells are not completely understood, and there is a need for ongoing monitoring to assess the durability of therapeutic response and the potential of disease relapse.549 Second, the risk of developing cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS) remain a concern, although it has been considered to be milder in the autoimmune setting as compared with that for treating cancers.550 Third, most CAR-T therapeutic modalities for treating SLE are autologous CD19-targeting CAR-T cells that are limited by the cell source and thus costly.550 To resolve these issues, several clinical trials investigating the feasibility of allogenic CD19-targeting CAR-T cells (NCT05988216, NCT06340490, NCT05859997, NCT06429800, NCT06294236, NCT06375993)561,562,563,564,565,566 and allogenic CD19-targeting CAR-NK cells (NCT06421701, NCT06010472)567,568 in treating SLE have been registered. A pre-clinical study successfully engineered Treg cells to express CD19-specific CAR using a mice lupus model through genetic editing, which yielded promising results.569 Rituximab, belonging to the first generation of CD20 antibodies, has been shown with clinical and histopathological evidence in improving the symptoms of LN patients570 and some SLE patients refractory to conventional therapies.495,571 In addition, rituximab decreased the anti-dsDNA antibody levels and increased the complement levels of SLE patients according to a phase III clinical trial involving 144 SLE patients (NCT00282347) and a phase II/III study of 257 participants (NCT00137969).572,573 Though rituximab has not been approval for treating SLE in the clinics, it has been recommended for refractory SLE management in 2023 EULAR recommendations.506 To further enhance the efficacy of rituximab in depleting B cells, two phase I trials (NCT06265220, NCT06581562) have combined ribuximab with AB-101, an allogenic NK cell product capable of killing target cells via antibody-dependent cell-mediated cytotoxicity (ADCC), in 2024.574,575 Other CD20 antibodies with clinical evidence for SLE treatment include, e.g., ofatumumab that decreased the disease activity of 3 juvenile SLE patients in a single center study,576 ocrelizumab that showed efficacy in treating LN patients in a phase III study (NCT00626197),577 and obinutuzumab that achieved increased renal responses and decreased flares among LN patients once combined with the standard therapies in a phase II study (NCT02550652).578,579 CD22, though being considered as an inhibitory receptor keeping the baseline level of B cell inhibition and the humoral immunity in check, has been used as a therapeutic target for depleting dysregulated B cells due to its restrictive expression on the surface of B cells.580 It has been shown that epratuzumab, a humanized anti-CD22 antibody, could effectively treat moderate and severe SLE patients with acceptable safety in several phase II and phase III studies (NCT00111306, NCT00383214, NCT00383513, NCT00624351).102,581 But epratuzumab failed in two phase III clinical trials (NCT01262365 NCT01261793).106 Abatacept, a soluble fusion protein linking the extracellular domain of human cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) to the modified Fc portion of human immunoglobulin G1, can bind to CD80/CD86 on the surface of APCs including B cells for impaired adaptive immune response activation and, in particular, B cell depletion.582 Thus, Abatacept has been associated with reduced production of anti-dsDNA antibody and proteinuria, and increased levels of C3 and C4 levels among active class III or IV LN patients according to a phase II/III clinical trial (NCT00430677).583,584,585,586 However, it showed only a limited therapeutic efficacy in treating non-life-threatening SLE in a phase II study (NCT00119678) (Table 2).585
PCs are differentiated B lymphocyte capable of antibody secretion.48,587 Instead of depleting B cells, one can also remove PCs. CD38 is a pleiotropic molecule expressed on the surface of PCs.48 Daratumumab, being an antibody targeting CD38, has led to a dramatic desirable therapeutic response in two refractory life-threatening SLE patients, with depleted PCs among other clinical indexes being observed.588 Several other clinical studies investigating the efficacy and safety of daratumumab as a SLE therapeutics have been currently registered and under investigations (NCT04868838, NCT04810754). Bortezomib, being a specific, reversible inhibitor of the 20S subunit of the proteasome,589 significantly reduced the amount of PCs in the peripheral blood and bone marrow by approximately 50%,590,591,592 and demonstrated its treatment efficacy in dealing with refractory and severe SLE (Table 2).589,593
Long-lived PCs are end-staged B cells residing in the bone marrow and other tissues that can survive for decades.594 Long-lived PCs may drive chronic immune responses due to its sustaining production of autoantibodies,595 leading to persistent SLE clinical syndrome or therapeutic resistance. It is worth noting that while removing long-lived PCs without affecting their precursors may not eradicate them, depleting B cells (the precursors of PCs) may promote the development of long-lived PCs, suggesting the need of a combinatorial therapeutic design. For instance, the small-molecule proteasome inhibitor bortezomib effectively depleted PCs and reduced autoantibodies but did not affect their precursors;596 rituximab, the chimeric antibody against the CD20 antigen, depleted B cells but stimulated the survival of long-lived PCs and thus led to rituximab resistance.597 Thus, combinatorial strategies targeting both long-lived PCs and their precursors such as the ‘bortezomib+rituximab’ regimen have been considered promising in SLE treatment.598
Similar to the rational of depleting B cells or PCs, depleting specific subsets of autoreactive T cells associated with SLE may also contribute to SLE attenuation. For instance, immunization of 6 SLE patients with inactivated autoreactive T cells decreased the disease activities without significant side effects being observed by inducing idiotype anti-idiotypic reactions.599
Increasing the immune tolerance of B cells may be another attractive strategy against SLE. Abetimus sodium is an immunomodulating agent capable of inducing the immune tolerance of B cells by directly binding to and cross-linking pathogenic autoantibodies such as anti-dsDNA antibody that are pathogenetic factors of LN (a chronic kidney disease developed accompanied with SLE patients).600 A phase III clinical trial has indicated that administrating abetimus sodium can effectively reduce the anti-dsDNA antibody levels in LN patients (NCT00035308).601 Intravenous injection of immunoglobulin to neutralize the overtly produced autoantibodies has been reported as an effective therapeutics for treating LN patients including refractory LN.602
Though the complement system may help recognize and clear the debris to avoid autoimmune response, over-activated complement system may lead to tissue damage towards exacerbated inflammation. Thus, attenuating over-activated complement system has been proposed to treat SLE in the clinical practice. For instance, eculizumab, a C5 antibody, has been shown effective in treating complement-mediated thrombotic microangiopathy among LN and SLE patients,603,604 with several successful cases being reported.605,606 Ravulizumab, another C5 antibody, has been registered to investigate its clinical efficacy in treating LN and IgA nephropathy (NCT04564339).607 Avacopan, a C5a receptor inhibitor, is currently being planned to be investigated for treating SLE.608 Pegcetacoplan, a C3 inhibitor, has been shown effective in treating complement-mediated nephropathy including LN in a phase II study (NCT03453619)609 (Table 2).
Current therapeutics restoring cytokine microenvironment homeostasis
To restore the balance of primary cytokines maintaining immune homeostasis in the microenvironment, cytokine-related therapeutics have been clinically used for SLE treatment. Specifically, administrating Th1-representative cytokines has been considered feasible for treating SLE. For instance, antibodies targeting receptors in response to Th2-generated cytokines represent an important portfolio of cytokine milieu modulating strategies for SLE treatment. For instance, IL6 promoted the maturation of B cells and differentiation of CD4+ T cells into Th17 but not Treg cells;610,611 and antibodies targeting the IL6 receptor such as tocilizumab, PF-04236921 and sirukumab showed desirable treatment efficacies in managing SLE. Specifically, the feasibility of using tocilizumab in treating SLE has been documented by a case report612 and a phase II study (NCT00046774);613 that of PF-04236921 has been reported by a post-hoc study of a phase II trial (NCT01405196);107 but that of sirukumab showed limited efficacies in treating CLE and SLE patients in a phase I study (NCT01702740)614 and among LN patients in a phase II trial (NCT01273389).615 It is also worth to mention that while favoring Th1 and Treg cells counterbalances the skewed immune microenviroment of SLE patients, it may revert the cytokine profile to the other end, rendering dual targeting of opposite cytokine axes towards microenvironment homeostasis an emerging trend for SLE therapeutics. For example, ustekinumab,614,615 a monoclonal antibody neutralizing the activities of IL12 (facilitating Th1 development) and IL23 (secreted by Th17 cells), has been show effective and safe in treating SLE from a two-year phase II study (NCT02349061).616,617 As another example, inhibitors of phosphodiesterase 4 (PDE4) such as apremilast have been shown effective in treating autoimmune diseases including refractory skin lesions of lupus in a trial involving 5 participants618 and in a phase II study involving 8 CLE patients (NCT00708916)619 by suppressing Th1 and Th17-mediated immune responses620 (Table 2).
Restoring Th17/Treg homeostasis via directly injecting cell materials has been proposed as a promising strategy for treating SLE. For instance, Tregs injected into lupus mice has attenuated the inflammatory response and alleviated the pathological syndromes of SLE.621 Stem cell therapies have shown a long-term efficacy and safety for SLE management622,623,624 by increasing the proportion of Treg cells among SLE patients.625 Low-dose IL2 has been associated with a rapid remission of SLE or CLE with good tolerance by rewiring the skewed Th1/Th2 cytokine microenvironment from several clinical trials of phase II or unavailable (NCT02955615, NCT02465580, NCT02932137, NCT02084238).626,627,628,629,630,631 Supply of IL10 antibody (that can be naturally produced by Treg cells) has been documented to be feasible in treating refractory SLE patients according to a clinical study involving 6 participants.632 From another point of view, secukinumab, an IL17A antibody functioning via blocking the interactions between IL17A and its receptor, has been successfully used to treat multiple autoimmune disorders such as ankylosing spondylitis, psoriasis, leprosy and,633,634 importantly, refractory LN patients.635,636,637 Pre-clinical studies have also shown that exosomes from umbilical cord blood MSCs can restore the Th17/Treg balance by lowering the percentage of Th17 subsets (Table 2).638
Some hormonal therapies have been shown effective in treating SLE via modulating the immuno-regulatory milieu. Glucocorticoids such as betamethasone,639 dexamethasone,640 hydrocortisone,641 prednisone,642 prednisolone,643 triamcinolone,644,645 methylprednisolone,393,646,647 which have long been applied topically or systematically for SLE treatment, are prone to inhibit the differentiation of Th17 cells and enhance that of Treg cells. In particular, glucocorticoids can decrease the number of Th17 cells and lower the level of IL17A for attenuated SLE symptom,648 and requires the aid of functioned Treg cells to exert anti-inflammation roles649 as evidenced by the upregulated Treg cells on glucocorticoid intake.650 Interestingly, increased levels of systemic glucocorticoid may protect the organism from running into Th1 over-dominance by modulating the Th1/Th2 balance, where the expression of Th1promoting cytokines such as IL12 and CSF were down-regulated and the secretion of Th2type cytokines such as IL4, IL10 and IL13 were up-regulated.651,652 Also, methylprednisolone pulse therapy has demonstrated its feasibility in treating SLE via promoting Treg cell differentiation (Table 2).647
Metabolic abnormalities remain as the underlying mechanism of Th17/Treg imbalance in SLE, as Th17 and Treg cells bear different metabolic patterns. In particular, while glycolysis, pentose phosphate pathway, fatty acid synthesis, and glutaminolysis are predominant in Th17 cells, fatty acid oxidation and oxidative phosphorylation are active in Treg cells. Thus, reprograming T cell metabolic patterns has emerged as an effective strategy for managing SLE patients through reversing the Treg/Th17 imbalance and restoring the immune homeostasis.653 Following this rational, rapamycin, a mTOR inhibitor, has been proposed for SLE treatment that functions by reprogramming T cell metabolic patterns towards prevented over-activation of Th17 cells and improved Treg proliferation, and been proven to alleviate disease severity among SLE patients in a phase I/II trial (NCT00779194).654,655 Statins, a class of drugs that inhibit cholesterol biosynthesis, have been reported capable of targeting Th17/Treg imbalance and alleviating Th17-mediated inflammatory response.656 For example, atorvastatin reduced atherosclerosis progression in lupus patients according to a phase III study (NCT00065806),657 and improved arterial stiffness and decreased SLE disease activity in an 8-week clinical study involving 37 SLE females;658 pravastatin decreased the total cholesterol and low density lipoprotein (LDL) levels in SLE patients;659 rosuvastatin reduced the lipid levels of SLE patients;660 and simvastatin decreased the levels of antiphospholipd antibodies among SLE patients.661 Besides, replacing glucose with galactose and supplementing 2-Deoxy-D-glucose (2-DG, an inhibitor of hexokinase that functions as the first rate-limiting enzyme of glycolysis) all diminished Th17 development and enhanced Treg differentiation78,79,80 (Table 2).
Current therapeutics rescuing impaired debris clearance machinery
SLE patients are characterized by high autoantibody loads that form ICs, inefficient clearance of which accelerate SLE syndroms. Most autoantibodies found in SLE carriers are IgG that can be recognized by FcγRIIIB, a type of Fcγ receptors capable of recognizing the Fc portion of IgG and expressed on neutrophils and a subset of basophils. FcγRIIIB participates in IC clearance by promoting phagocytosis, adhesion, and the respiratory burst in neutrophils.662 FcγRIIB is another member of the FcγR family that are primarily expressed on B cells, macrophages, and DCs.663 As dysfunctional FcγRIIB and FcγRIIIB have both been implicated in priming SLE,662 therapeutics mimicking the effect of these receptors have been established for SLE management. For instance, valziflocept, a recombinant soluble human FcγRIIb that binds to ICs as a decoy, has been shown promising in a double-blind placebo-controlled multicenter study for managing SLE symptoms.663,664
Purified or recombinant complement proteins may not be recommended for treating SLE in the clinics due to the possible damaging effects of overtly stimulated complement system. Specifically, hyperactivated complement system via the alternative pathway may damage cells such as podocytes and tissues such as the blood vessel endothelium that deteriorate the SLE symptoms. Thus, the fresh frozen plasma containing multiple complement proteins has been launched in the clinics, with therapeutic successes being documented in treating C1q-deficient SLE patients665 and those harboring C2 deficiency.666 Plasma exchange may represent another strategy to fuel sufficient complements in the blood to remove pathological antibodies and their complexes with the immune components in time for alleviated SLE symptoms (Table 2).667,668,669
Conclusions
We reviewed the history of the SLE research field and the epidemiology of this disease, grouped the mechanisms-of-action driving SLE pathogenesis into three categories, i.e., activating the immune response, skewing the cytokine microenvironment, and impairing the debris clearance machinery; summarized current knowledge on SLE diagnosis by the disease onset, activity and comorbidities; identified risk factors predisposing SLE at the genetic, epigenetic, hormonal, extrinsic levels; and, importantly, classified current SLE preventive and treatment strategies following the logic of the identified mechanisms. It is worth noting that these groups used for classifying current SLE preventive and therapeutic strategies are not mutually exclusive, as most SLE management approaches take action through multiple mechanisms. For instance, glucocorticoid, primarily functions via modulating the cytokine microenvironment,648,649,650,651,652 also attenuates the immune response by inhibiting the maturation and activity of DCs, interfering with TCR signaling, and inducing B cell apoptosis as well as affecting the downstream pathways of B cell receptor signaling such as NF-κB.349 As another example, the methylprednisolone pulse therapy functions not only by promoting Treg cell differentiation, but also via inducing the apoptosis of CD4+ T cells.647 Similarly, hydroxychloroquine, known capable of alleviating SLE and largely by blocking TLR signaling,455 also modulates the cytokine distribution and homeostasis among Th1, Th2, Th17 and Treg cells.670,671
Given our incremental understanding on the priming role of intrinsic features in predisposing SLE and the increasing need for precision medicine, focusing on personalized SLE treatment taking advantages of genetic or immunological markers may represent a promising therapeutic perspective. Thus, identifying representative markers associated with each of the three identified pathogenesis stages and establishing rapid yet cost-effective screening techniques may aid in the therapeutic design and lead one future direction.





Responses