Fine-tuning of dopamine receptor signaling with aripiprazole counteracts ketamine’s dissociative action, but not its antidepressant effect

Introduction
The rapid antidepressant and antisuicidal effects of ketamine, previously used as an intravenous anesthetic, are well documented [1, 2]. The use of ketamine in the treatment of treatment-resistant depression continues to expand, particularly in the USA. Off-label use of intravenous ketamine for depression is widespread in US clinics [3]. In 2019, intranasal esketamine (SPRAVATO, Janssen Pharmaceuticals), which contains only the (S)-(+)-enantiomer of ketamine, received FDA approval for adjunctive use in treatment-resistant depression. However, ketamine’s propensity to induce dissociative symptoms and associated discomfort and fear after administration is an important clinical concern, necessitating the observation of symptoms during and after administration. Therefore, even when using intranasal formulations, its administration is limited to healthcare facilities (SPRAVATO product monograph).
The presence and intensity of dissociative symptoms following ketamine administration appear to be uncorrelated with its antidepressant effects [4, 5]. The adverse symptoms of ketamine are similar to those of schizophrenia [6]. In addition, several adjunctive treatments with antipsychotics that antagonize the dopamine D2 receptor (D2R) have been investigated to mitigate these symptoms, including haloperidol [7] and risperidone [8] in animal studies. A concern of the adjunctive use of antipsychotics was raised by an animal study by Li et al. showing that D2R antagonism weakens the antidepressant effect of ketamine [9]. However, no controlled studies have examined whether adjunctive antipsychotics attenuate the antidepressant effect of ketamine in humans, although there are observational studies and case reports in which some subjects treated with ketamine took various antipsychotics concomitantly [10,11,12,13]. Moreover, the mechanisms of the drug interactions have not been investigated.
In the study by Li et al., the antipsychotic haloperidol, a D2/D3 receptor antagonist, suppressed not only ketamine’s psychotomimetic effect, but also its antidepressant effect [9]. However, it was difficult to discriminate between these actions of haloperidol because both behavioral tests, which are locomotor activity-dependent, were conducted at the same time when ketamine increased locomotor activity.
In the present study, we investigated whether D2/D3 receptor modulation might be an effective strategy for counteracting the psychotomimetic action of ketamine, while preserving its antidepressant effects. To this end, we evaluated the adjunctive efficacies of raclopride and aripiprazole, a D2/D3 receptor antagonist and a partial agonist, respectively, on the antidepressant-like effect of ketamine in the forced swim test (FST). Furthermore, we perform brain-wide Fos mapping to identify the brain regions mediating the differential effects of raclopride and aripiprazole on ketamine’s actions. Lastly, to validate our preclinical findings, we conduct an exploratory clinical research study on the effect of aripiprazole on dissociative symptoms in depressive patients treated with ketamine. Based on our findings, we propose that the adjunctive use of aripiprazole might improve the safety of ketamine therapy for treatment-resistant depression.
Results
Aripiprazole suppresses the psychotomimetic effects of ketamine, but not its antidepressant-like action
Haloperidol, a non-selective D2/D3 receptor antagonist, is reported to prevent the antidepressant-like effect of ketamine [9]. However, they examined the effect of haloperidol on ketamine’s antidepressant-like effect using the FST, 30 min after ketamine injection, when ketamine-induced hyperlocomotion occurs [7] (Supplementary Fig. 1) and it can influence immobility time in the FST. To exclude the possibility that D2/D3 receptor antagonism impacts immobility time by affecting locomotion, we evaluated the effects of D2/D3 receptor modulators on ketamine’s antidepressant-like effect 24 h after administration of the drugs. First, we examined the effective dose range of a selective D2/D3 antagonist, raclopride, on ketamine-induced hyperlocomotion, which is related to its psychotomimetic effect, by quantifying locomotor activity for 40 min after ketamine injection in the open field test (OFT). Co-administration of raclopride with ketamine reduced ketamine-induced hyperlocomotion in a dose-dependent manner at a dose range of 0.1–1.0 mg/kg (Fig. 1a). Next, to assess the effects of raclopride on the antidepressive effect of ketamine, we performed the FST 24 h after drug administration. While ketamine-treated mice showed a reduction in immobility time compared with vehicle-treated mice, the mice co-treated with 0.1 mg/kg raclopride and ketamine did not show a decrease in immobility time (Fig. 1b, c), suggesting that antagonizing D2/D3 receptors suppresses both the psychotomimetic and antidepressant effects of ketamine.

a Inhibitory effect of raclopride on ketamine-induced hyperactivation (vehicle: n = 11 mice; raclopride 0.1 mg/kg: n = 8 mice; raclopride 0.2 mg/kg: n = 8 mice; raclopride 0.3 mg/kg: n = 3 mice; raclopride 1.0 mg/kg: n = 3 mice; Dunnett’s multiple comparisons test, ****P < 0.0001). b, c Total immobility time and the time course in the FST (vehicle: n = 18 mice; ketamine: n = 17 mice; raclopride 0.1 mg/kg plus ketamine: n = 18 mice; Dunnett’s multiple comparisons test, *P < 0.05 vs vehicle in (b). Two-way repeated-measures ANOVA followed by uncorrected Fisher’s LSD post hoc test, *P < 0.05 vs vehicle in (c)). d Suppressive effect of aripiprazole on normalized ketamine-induced locomotion (vehicle: n = 11; aripiprazole 0.1 mg/kg: n = 6; aripiprazole 0.3 mg/kg: n = 8; aripiprazole 1.0 mg/kg: n = 6; Dunnett’s multiple comparisons test, *P < 0.05, **P < 0.01). e, f Total immobility time and the time course in the FST 1 day after administration (vehicle: n = 24 mice; ketamine: n = 23 mice; aripiprazole 1.0 mg/kg plus ketamine: n = 24 mice; Dunnett’s multiple comparisons test, ***P < 0.001, **** P < 0.0001 vs vehicle in (e). Two-way repeated-measures ANOVA followed by uncorrected Fisher’s LSD post hoc test, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs vehicle, # P < 0.05 vs ketamine in (f)).
Next, we examined the effect of aripiprazole, a D2/D3 receptor partial agonist, on ketamine’s actions. In the OFT, aripiprazole treatment 15 min before ketamine injection reduced ketamine-induced hyperlocomotion in a dose-dependent manner at a dose range of 0.3–1.0 mg/kg, and the efficacy of the 1.0 mg/kg dose was comparable to that of 0.1 mg/kg raclopride (Fig. 1d). Therefore, we examined the effect of the co-administration of 1.0 mg/kg aripiprazole and ketamine using the FST, which revealed significantly decreased immobility time (Fig. 1e, f). Interestingly, co-treatment with aripiprazole augmented the antidepressant-like effect of ketamine (Fig. 1f). These results suggest that the D2/D3 receptor partial agonistic activity of aripiprazole selectively attenuates the psychotomimetic effect of ketamine, while preserving its antidepressive effect.
Comparative Fos mapping of neuronal activity
To identify the brain regions involved in the modulation of ketamine’s effects by raclopride and aripiprazole, we conducted brain-wide Fos mapping to compare neuronal activity changes induced by different combinations of these drugs, 105 min after ketamine injection. Because D2R antagonism in the striatum is associated with suppression of the positive symptoms of schizophrenia [14], we first counted Fos-positive (Fos+) cells among D2R-positive cells in the caudate putamen (Cpu), located in the dorsal striatum. Notably, both aripiprazole and raclopride significantly increased Fos+ cells among the D2R-positive cell population in the Cpu (Fig. 2a, b), suggesting that both the D2/D3 antagonist and the partial agonist suppress the psychotomimetic effect of ketamine through mechanisms similar to those by which antipsychotics suppress the positive symptoms of schizophrenia.
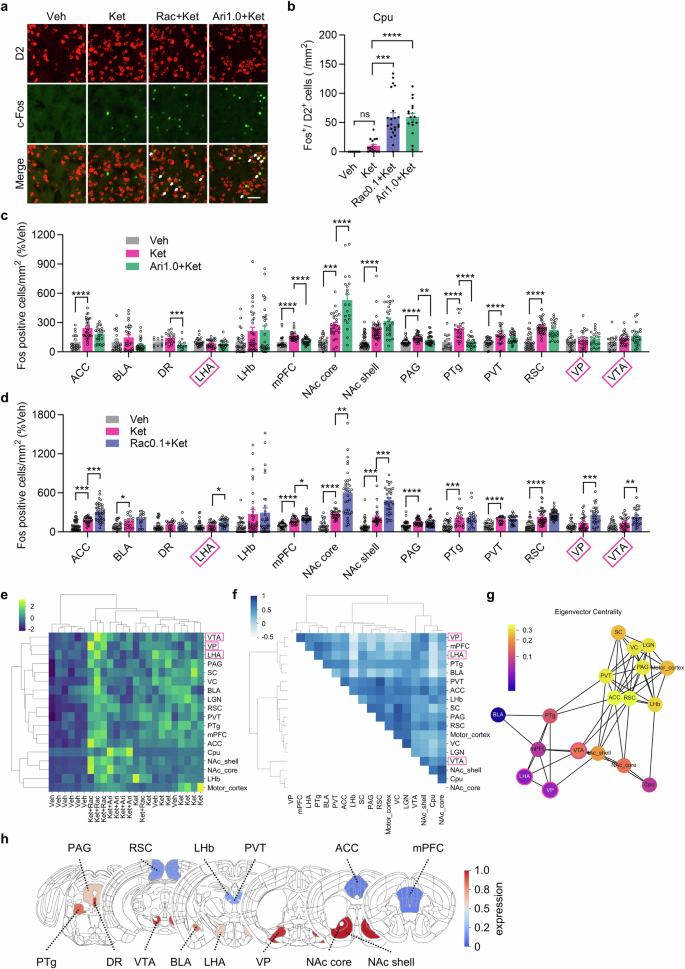
a, b Representative images showing Fos and D2R immunoreactivity in the Cpu, 105 min after administration of vehicle, ketamine, raclopride plus ketamine or aripiprazole plus ketamine (scale bar is 50 μm, allows indicate Fos/D2 double-positive cells), as well as the number of Fos/D2R-double-positive cells in the Cpu (vehicle: n = 7 slices from 2 mice; ketamine: n = 17 slices from 5 mice; raclopride plus ketamine: n = 22 slices from 5 mice; aripiprazole plus ketamine: n = 18 slices from 5 mice). Dunn’s multiple comparisons test, ***P < 0.001, ****P < 0.0001. c Quantification of Fos immunostaining in the various brain regions associated with antidepressant effects 105 min after administration of vehicle (gray, n = 10–46 slices from 4 mice), ketamine (red, n = 18–43 slices from 4 mice) or aripiprazole plus ketamine (green, n = 14–38 slices from 4 mice). Dunn’s multiple comparisons test or Dunnett’s multiple comparisons test was used depending on normality. **P < 0.01, ***P < 0.001, ****P < 0.0001. d Quantification of Fos immunostaining in multiple brain regions associated with antidepressant effects 105 min after administration of vehicle (gray, n = 22–42 slices from 4 mice), ketamine (red, n = 18–40 slices from 4 mice) or raclopride plus ketamine (blue, n = 19–46 slices from 4 mice). Dunn’s multiple comparisons test or Dunnett’s multiple comparisons test was used depending on normality. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. e Hierarchical clustering of mice and brain regions by Fos+ cell density. f Correlation matrix of Fos+ cell densities between pairs of brain regions. g Network consisting of the nodes of brain regions and connections of Pearson’s r with the absolute value > 0.5. The color scale shows eigenvector centrality. h D2R mRNA expression map in depression-related brain regions from Allen brain map dataset (https://portal.brain-map.org/). ACC anterior cingulate cortex, BLA basolateral amygdaloid nucleus, DR dorsal raphe nucleus, LHA lateral hypothalamus, LHb lateral habenula, mPFC medial prefrontal cortex, NAc nucleus accumbens, PAG periaqueductal gray, PTg pedunculotegmental nucleus, PVT paraventricular nucleus of the thalamus, RSC retrosplenial cortex, VP ventral pallidum, VTA ventral tegmental area.
Next, we quantified Fos+ cells in the brain areas previously reported to be associated with depressive symptoms [15]. Two mechanisms whereby raclopride attenuates ketamine’s antidepressant effect are plausible: (A) raclopride inhibits activation of brain regions involved in ameliorating depressive symptoms; (B) raclopride activates brain regions that exacerbate depressive symptoms. Given that D2/D3 receptors are coupled to Gi proteins to inhibit membrane excitability, we postulated that mechanism (B) is more likely. In this mechanism, antagonizing D2/D3 receptors with raclopride would disinhibit neuronal activity. Indeed, there was no brain region where the combination of raclopride and ketamine showed fewer Fos+ cells than ketamine only or its combination with aripiprazole (Fig. 2c, d). Rather, only the combination of raclopride and ketamine significantly increased Fos+ cells in the lateral hypothalamic nucleus (LHA), ventral pallidum (VP) and ventral tegmental area (VTA), further supporting mechanism (B). Using the data of Fos+ cell density, we conducted unsupervised hierarchical clustering of the treated mice, showing a reasonable separation of the mice by the treatments (Fig. 2e). The cluster of the mice treated with Ket+Ari was closer to that of ketamine-treated mice than that of the mice treated with Ket+Rac, in line with the behavioral outcomes in the FST. In the brain region clustering, LHA, VP and VTA, selectively activated by Ket+Rac, are closely clustered. Next, we analyzed the correlations of neural activations between pairs of the brain regions and found the inter-connected brain regions, such as NAc core, Cpu, NAc shell and VTA, were closely clustered (Fig. 2f), implying the Fos+ cell density correlation links with functional connectivity. To identify the hub brain region among those selectively activated by Ket+Rac, we constructed a network where the brain regions and the correlations with |r|> 0.5 are the nodes and the connections, respectively (Fig. 2g). Among the brain regions selectively activated by Ket+Rac, the VTA showed the highest eigenvector centrality, meaning it had the most connections with high-impact brain regions. In the VTA, D2Rs are highly expressed (Fig. 2h) [16], and systemic injection of a D2/D3 antagonist was reported to induce phasic activation of TH-positive neurons in the mouse social defeat stress (SDS) model [17] as well as depressive symptoms [18, 19]. Taken together, we hypothesized that the activation of the VTA by the D2/D3 antagonist would be involved in abolishing the antidepressant effect of ketamine.
Activation of dopaminergic neurons in the VTA attenuates ketamine’s antidepressive effect
To clarify the role of VTA activation in depressive symptoms, we used the SDS model. Raclopride was locally infused into the VTA of SDS mice, immediately followed by intraperitoneal administration of ketamine, and the following day, sociability was assessed using the social interaction test (SIT) (Fig. 3a). In the SIT, intra-VTA infusion of aCSF together with ketamine i.p. injection improved social interaction ratios in SDS mice compared with those before the treatment, whereas intra-VTA aCSF infusion together with saline i.p. injection didn’t. In comparison, intra-VTA infusion of raclopride with ketamine i.p. injection did not improve social interaction ratios (Fig. 3b, c). Next, to examine dopaminergic neuron activation in the VTA, we quantified Fos/TH-double-positive cells in the VTA of treated mice. While intra-VTA infusion of aCSF together with ketamine i.p. did not change the number of Fos/TH-double-positive cells compared with vehicle, intra-VTA infusion of raclopride together with ketamine i.p. injection significantly increased the number of Fos/TH-double-positive cells 105 min after administration (Fig. 3d, e). Taken together, these findings suggest that raclopride suppresses the antidepressant effect of ketamine by activating VTA dopaminergic neurons.
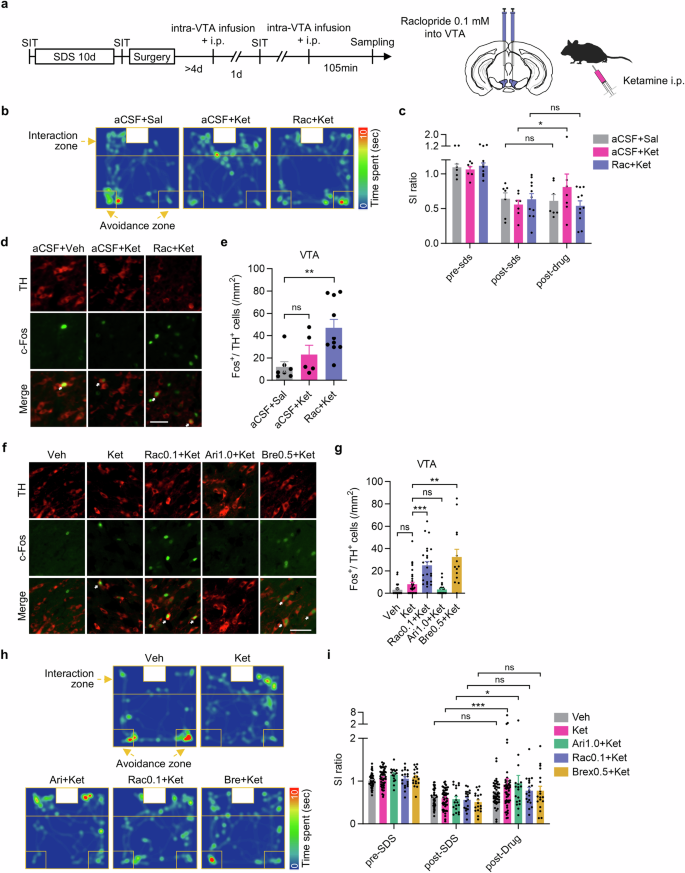
a Experimental procedures for social defeat stress (SDS), intra-VTA infusion and the social interaction test (SIT). b Representative behavioral traces 1 day after intra-VTA infusion of aCSF plus saline i.p., intra-VTA aCSF plus ketamine i.p. or intra-VTA raclopride plus ketamine i.p. in the SIT. c SI ratio in the SIT (aCSF + Sal: n = 7 mice; sCSF + Ket: n = 7 mice; Rac + Ket: n = 11 mice; mean ± s.e.m.; two-way repeated-measures ANOVA followed by Fisher’s LSD test, *P < 0.05). d Representative immunoreactivities for TH and Fos in the VTA of mice administered intra-VTA aCSF and saline i.p., intra-VTA aCSF and ketamine i.p., or intra-VTA raclopride and ketamine i.p., (scale bar is 50μm, allows indicate Fos/TH double-positive cells). e Quantitation of TH/Fos-double-positive cells in the VTA (aCSF + Sal: n = 7 slices from 3 mice; aCSF + Ket: n = 5 slices from 3 mice; Rac + Ket: n = 10 slices from 3 mice; Dunn’s multiple comparisons test, **P < 0.01). f, g Representative images of Fos and TH immunoreactivities in the VTA 105 min after the indicated i.p. treatments (scale bar is 50 μm, allows indicate Fos/TH double-positive cells), and the number of Fos/TH-double-positive cells (vehicle: n = 38 slices from 6 mice; ketamine: n = 35 slices from 6 mice; raclopride plus ketamine: n = 25 slices from 4 mice; aripiprazole plus ketamine: n = 24 slices from 4 mice; brexpiprazole plus ketamine: n = 14 slices from 3 mice; Dunn’s multiple comparisons test, **P < 0.01, ***P < 0.001). h Representative behavioral traces in the SIT, 1 day after i.p. administration of the indicated drugs. i Social interaction ratio in the SIT (vehicle: n = 47 mice; ketamine: n = 51 mice; aripiprazole plus ketamine: n = 18 mice; raclopride plus ketamine: n = 19 mice; brexpiprazole plus ketamine: n = 18 mice; two-way repeated-measures ANOVA followed by Dunnett’s multiple comparisons test, *P < 0.05, ***P < 0.001).
Next, we evaluated the effects of D2/D3 receptor partial agonists, aripiprazole and brexpiprazole, on dopaminergic neuron activity in the VTA, when combined with ketamine (Fig. 3f, g). In line with the above experiments, systemic co-administration of raclopride and ketamine increased the number of Fos+ VTA dopaminergic neurons. In contrast, co-treatment with aripiprazole and ketamine did not increase the number of Fos/TH-double-positive cells compared with vehicle and ketamine. Surprisingly, the combination of 0.5 mg/kg brexpiprazole (Supplementary Fig. 3) and ketamine increased Fos+ cell counts among the TH-positive population in the VTA, as co-treatment with raclopride and ketamine did. This suggests that due to a 10–20% lower Emax for D2/D3 receptors than aripiprazole, brexpiprazole works as an effective D2/D3 antagonist rather than a partial agonist in the VTA [20].
Next, we examined the effects of those drug combinations on depressive-like symptoms in SDS mice using the SIT. In line with the Fos results in the VTA, only the combination of aripiprazole and ketamine improved the social interaction ratio as ketamine single treatment did, whereas neither brexpiprazole and ketamine nor raclopride and ketamine affected the social interaction ratio (Fig. 3h, i). These results suggest that when combined with ketamine, only aripiprazole, among antipsychotics with affinity for D2/D3 receptors, preserves the antidepressant effect of ketamine by circumventing the activation of dopaminergic neurons in the VTA through finely modulating D2/D3 receptors.
Aripiprazole suppresses dissociation in depressed patients treated with ketamine
The above findings in mice led us to examine the effect of aripiprazole in depressed patients treated with ketamine. Among the nine recruited cases, eight subjects completed the whole treatment (Supplementary Fig. 5). One participant in the analysis opted not to take subsequent ketamine doses due to nausea following the initial dose, resulting in study dropout. The data from this case were incorporated exclusively in the analysis involving comparison of symptoms before and after the first dose. One case among eight who completed the whole schedule was excluded from the analysis because the Clinician-Administered Dissociative States Scale (CADSS) score was 30 before the initial ketamine administration, which was deemed a clinically significant indicator that warranted exclusion. The average CADSS values were increased after each ketamine administration, except for the third administration following the intake of aripiprazole, 12 mg (Fig. 4b, Supplementary Table 1). Also, the percentage of positive cases with dissociative symptoms in the post-ketamine assessments was the lowest (14%) in the third session (Fig. 4c). On the relationship between aripiprazole blood concentrations and dissociative symptoms, there was a significant difference in the proportion of positive cases with dissociative symptoms between the group with aripiprazole blood concentrations 20 ng/ml or higher and the group with concentrations below the threshold (Fig. 4d). A blood level of 20 ng/mL aripiprazole was achieved by a single dose of 12 mg, but not 3 mg. The mean Montgomery–Åsberg depression rating scale (MADRS) scores showed significant reductions (in comparison with pre-treatment in the first session) after the third and fourth treatments, as well as before and after the fifth treatment (Fig. 4e, Supplementary Table 2). The results of the other evaluations conducted in this clinical study are available in the supplemental information (Supplementary Figs. 6–9).
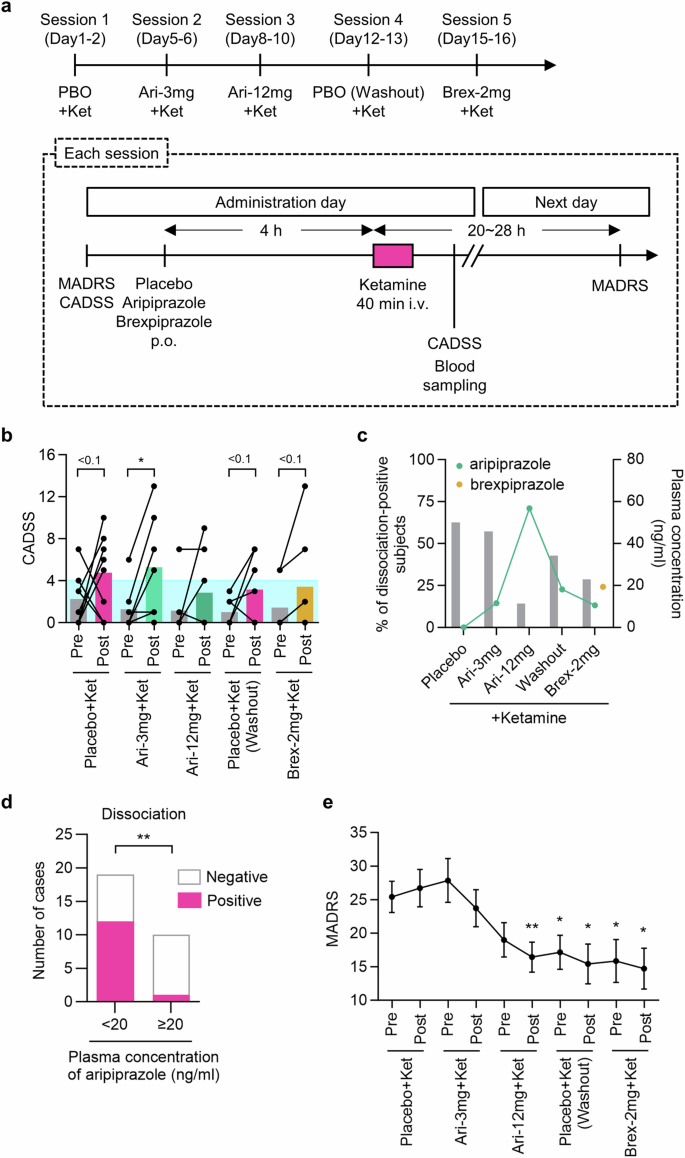
a Study schedule and the procedure for assessment of the effects of ketamine, aripiprazole plus ketamine, and brexpiprazole plus ketamine. b Changes in dissociative symptoms as assessed with the CADSS after administration of ketamine (One-tailed Wilcoxon matched-pairs signed rank test, *P < 0.05, P < 0.1). The blue shade indicates the normal range, < 4. c Percentage of dissociation-positive cases (post-ketamine CADSS > 4 and CADSS change >0) and mean plasma concentrations of antipsychotics at each dosing time (n = 7, except n = 8 for percentage of dissociation-positive cases at placebo treatment). d The number of dissociation-positive cases when the concentration of aripiprazole in plasma was < or ≥ 20 ng/mL in sessions one to four (n = 29 cases, Fisher’s exact test, **P < 0.01). e Changes in the depression symptoms assessed with the MADRS throughout treatment (n = 7, one-way repeated-measures ANOVA, treatment, F2,15 = 15.00, P = 0.0001; Dunnett’s multiple comparisons test: *P < 0.05, **P < 0.01, vs before treatment with placebo plus ketamine). MADRS: Montgomery–Åsberg depression rating scale, CADSS: Clinician-Administered Dissociative States Scale.
Discussion
Here, we investigated various combinations of drugs, with the aim of counteracting the psychotomimetic effect of ketamine, while preserving its antidepressant action. Although several previous studies examined the effects of antipsychotics on psychotomimetic behaviors induced by ketamine in rodents, their effects on the antidepressant action of ketamine remained unclear. The mechanisms of action of antipsychotics converge on D2R antagonism [21]. Therefore, we used raclopride, a D2/D3 receptor selective antagonist [22], as a representative antipsychotic. The effective doses of raclopride and aripiprazole for suppressing psychotomimetic symptoms induced by ketamine were 0.1 mg/kg and 1.0 mg/kg, respectively, based on the OFT, in which they decreased ketamine-induced hyperlocomotion by about 80%. Intraperitoneal injection of aripiprazole at 1.0 mg/kg in mice is postulated to lead to 80% occupancy of D2Rs [23], which is comparable to the level achieved by clinically effective doses of aripiprazole (12–24 mg) in humans. Those doses of raclopride and aripiprazole were evaluated for their effects on the antidepressant-like action of ketamine in the FST, 1 day after ketamine injection, when hyperlocomotion induced by ketamine completely disappeared. Co-treatment of raclopride and ketamine eliminated the antidepressant-like effect of ketamine, suggesting that D2/D3 receptor antagonists inhibit ketamine’s antidepressive effect, in accordance with a previous study on haloperidol [9]. Notably, co-treatment of aripiprazole and ketamine preserved and even enhanced the antidepressant-like effect of ketamine in the FST. These findings suggest that D2/D3 receptors agonism in specific brain regions is necessary for ketamine’s antidepressive effect and additional agonism further facilitates neural activity changes underlying the antidepressant action.
To uncover the underlying mechanisms of action, we performed brain-wide Fos mapping to quantify neuronal activity changes in the various brain regions after the treatments. The activation of D2/D3 receptors, which are coupled to Gi proteins, leads to the cell-autonomous suppression of neuronal activity [24]. In contrast, antagonizing these receptors enhances neuronal activity in brain regions where dopamine levels are high enough to sustain D2/D3 receptor activation under basal conditions. Indeed, raclopride co-treatment with ketamine induced more widespread neuronal activation than ketamine alone, whereas co-treatment with aripiprazole, a D2/D3 partial agonist, narrowed the activated brain areas. In the Cpu, D2 cells were activated by both the raclopride plus ketamine and aripiprazole plus ketamine co-treatments. Given that antagonizing D2Rs in the striatum, which includes the Cpu, is critical for suppressing psychosis in schizophrenia [25, 26], ketamine-induced psychotomimetic symptoms may, at least partially, involve mechanisms similar to those underlying psychosis.
We observed two types of brain regions, which differed in their responses to raclopride and aripiprazole co-treatments with ketamine. The first is a region activated by raclopride but not by aripiprazole, and the second is a region not affected by raclopride, but suppressed by aripiprazole. We postulate that the first type of brain region might participate in the raclopride-induced attenuation of ketamine’s antidepressant-like effect, while the second may be involved in the aripiprazole-induced augmentation of ketamine’s antidepressive action. We demonstrated that the activation of dopaminergic neurons in the VTA, representing the first type of brain region, was crucial for the raclopride-induced attenuation of ketamine’s antidepressive effect. Although we did not investigate the role of the second type of brain region, including the mPFC, in the aripiprazole-induced augmentation of ketamine’s antidepressant-like effect, previous reports imply that suppressing neuronal activation in the mPFC with aripiprazole might augment the antidepressive action of ketamine. Optogenetic activation of D2 cells in the mPFC reduces social exploration time [27], and conversely, chemogenetic suppression of D2 cells in the mPFC of ketamine-treated mice reduces the immobility time compared with mice treated with ketamine alone [28]. These observations are consistent with our finding that aripiprazole reduced Fos+ D2 cells in the mPFC (Supplementary Fig. 4) and enhanced ketamine’s antidepressant effect. Taken together, the findings suggest that ketamine causes widespread brain activation, leading to both the suppression and enhancement of depressive phenotypes, and that aripiprazole preferentially suppresses the latter, resulting in the enhancement of the antidepressive effect. The roles of pro-depressive brain regions such as mPFC in the augmentation of ketamine’s antidepressive action by aripiprazole warrant future study. Their mechanism of action would be distinct from those of enhancing the effects of conventional antidepressants such as SSRIs by antipsychotics including brexpiprazole and D2R antagonists.
A causative role of VTA activation in the attenuation of ketamine’s antidepressant-like effect was clearly demonstrated in SDS mice, the most well-established animal model of depressive disorders. Brexpiprazole, which increased dopaminergic neuronal activation in the VTA as raclopride did, attenuated ketamine’s antidepressant-like effect when co-administered in SDS mice, whereas aripiprazole, which did not activate dopaminergic neurons in the VTA, preserved ketamine’s antidepressive effect. These results provide strong evidence of a key role of dopaminergic neuron activation in the VTA in the attenuation of ketamine’s antidepressant-like effect.
The role of the VTA in depressive symptoms is complex, and the phenotype is highly dependent on context, neuronal activity and projections [15]. Specifically, phasic activation of VTA dopaminergic neurons in the VTA–NAc circuit causes a depressive-like phenotype [19]. Moreover, systemic administration of raclopride induces phasic activation of VTA dopaminergic neurons [17]. VTA dopaminergic neurons showing phasic activation project to the shell of the NAc [29]. Thus, raclopride may attenuate the antidepressive effect of ketamine by inducing phasic activation of the VTA–NAc circuit. Interestingly, although both brexpiprazole and aripiprazole are approved by FDA as adjunctive therapy for MDD, and they are similar in chemical structure and pharmacological properties, their adjunctive effects on the antidepressive action of ketamine are highly divergent. Their contrasting effects might be attributed to differences in the Emax for D2Rs, which are 43 and 61% for brexpriprazole and aripiprazole, respectively [20]. The lower Emax of brexpiprazole could make it function as a D2R antagonist in the VTA and inhibit Gi signaling, resulting in D2 neuron activation. Conversely, aripiprazole, with a higher Emax, may function as a D2R agonist in the VTA to activate Gi signaling, thereby preventing D2 cell activation. The Emax of aripiprazole is well suited to antagonizing D2Rs in the Cpu, which has a high dopamine concentration, but not in the VTA. Such fine tuning is unlikely to be achieved by antipsychotics that antagonize D2Rs, and these would therefore likely suppress both the antidepressive and psychotomimetic effects of ketamine, as raclopride did. The only exception is cariprazine, which is a D2R partial agonist with an Emax comparable to that of aripiprazole [30]. Therefore, cariprazine might also counteract ketamine-induced dissociation, but not its antidepressant effect. However, we cannot exclude the possibility that the pharmacological effects of antipsychotics on receptors/transporters other than D2Rs may influence the antidepressant effect of ketamine. The limitation of our preclinical study is that only male animals were used. Therefore, the generalizability of our findings to female animals needs to be investigated in future work.
In our clinical study, premedication with 12 mg aripiprazole reduced dissociative symptoms after ketamine administration. In contrast, brexpiprazole did not suppress dissociative symptoms. Previous clinical studies show that, in healthy subjects, haloperidol does not reduce ketamine-induced dissociative symptoms [31, 32] and, in schizophrenic patients, clozapine blunts the ketamine-induced increase in positive symptoms [33]. Given that our Fos mapping showed distinct effects of the D2R antagonist and the partial agonist in several cortical regions (Supplementary Fig. 2), different antipsychotics may differentially modulate ketamine-induced psychosis through differential effects on neural activity, depending on their modes of actions on D2Rs and other receptors.
This study demonstrates, for the first time, an association between ketamine-induced dissociative symptoms and blood levels of antipsychotic drugs. The optimal concentration of aripiprazole for schizophrenia is 120–270 ng/ml [34]. In the present study, dissociative symptoms were significantly reduced when blood concentrations of aripiprazole reached 20 ng/ml or higher. This suggests that the blood levels of antipsychotics required to suppress dissociative symptoms induced by ketamine are well below the levels needed to alleviate psychiatric symptoms in schizophrenia. As for brexpiprazole, the relationship between blood concentration and dissociative symptoms could not be examined, because aripiprazole administered in the third session persisted into the fifth session (brexpiprazole administration) (Fig. 4c).
It is important to show that ketamine’s antidepressant effect is not inhibited by concomitant use of antipsychotics. In this clinical study, a total of five doses of ketamine, including two doses with aripiprazole and one dose with brexpiprazole, led to a 42% reduction in the total MADRS score in an uncontrolled observation, which is similar to that seen in previous studies of repeated ketamine injections [35]. While there is no report directly examining whether concomitant use of aripiprazole or brexpiprazole affects the antidepressant effects of ketamine in humans, the concurrent use of some antipsychotics was associated with non-responsiveness to ketamine in an observational study [13]. In our exploratory analysis of MADRS score changes between pre- and post-treatments in each session, the MADRS scores were more decreased by co-administration of aripiprazole with ketamine than ketamine alone (Supplementary Fig. 9), consistent with our preclinical finding (Fig. 1f).
The limitations of our current clinical study include the small sample size and the non-randomized timing of antipsychotic administration. The primary aim was to investigate the effect of aripiprazole in suppressing dissociative symptoms, and therefore, no randomized comparison of its antidepressant effects was conducted. The effect of concomitant aripiprazole on ketamine’s antidepressant effect needs to be evaluated in a larger randomized, controlled, clinical study.
In conclusion, our preclinical and clinical studies suggest that the combined use of aripiprazole and ketamine may be therapeutically useful for treatment-resistant depression. Furthermore, our findings provide insight into the substrates and neural mechanisms of the pharmacological interactions between ketamine and different classes of antipsychotics.
Methods
Study approval
All experimental procedures involving animals were approved by the Institutional Animal Care and Use Committees of Kyoto University. The clinical study with human subjects was approved by the Committee on Medical Ethics of Kyoto University (Y0058), registered with the Japan Registry of Clinical Trials (jRCTs051210192, “Efficacy and Safety study of the therapeutic approach using ketamine in refractory depression.”), and adhered to the principles of the 1964 Declaration of Helsinki and its later amendments. All participants provided written informed consent to participate in the study.
Preclinical study
Animals
Our study examined male mice because the male is postulated to exhibit less variability in behavioral assays. C57BL/6 J mice (B6 mice, CLEA Japan, Inc, Japan) were used in all experiments. In addition, retired breeder Jcl:ICR mice (ICR mice, CLEA Japan, Inc, Japan) were used as aggressors in the social defeat stress procedure and in the social interaction test (SIT). Mice were maintained under a strict 12-h light-dark cycle (light on at 8:00 a.m. and off at 8:00 p.m.), with mouse chow and water provided ad libitum. The number of animals used for each experiment was determined referring to the previous studies [36,37,38]. The animals were randomized by body weight or SI ratio (SDS mice).
SDS
For CSDS [39], retired male breeder ICR mice were used as aggressors. ICR mice were housed in the social defeat cage separated by a clear perforated Plexiglas divider 24 h before exposure to the first stressor. C57BL/6 J mice were exposed to an aggressor for 10 min on 10 consecutive days and housed on the opposite side of the divider after the social defeat stress for a 24-h period. C57BL/6 J mice were exposed to a novel aggressor for each daily defeat session.
Intraperitoneal administration
Ketamine (Daiichi Sankyo Propharma, Japan) and raclopride (1810, Tocris Bioscience, UK) were dissolved in saline. Aripiprazole (013-23833, Wako Pure Chemical Industries, Japan) and brexpiprazole (HY-15780, MedChemExpress, USA) were dissolved in 5% Tween 80 (P8074, Sigma-Aldrich, USA) in saline. The drugs were intraperitoneally injected (10 ml/kg body weight). Raclopride and ketamine were simultaneously injected. Aripiprazole and brexpiprazole were injected 15 min before ketamine administration.
Intra-VTA infusion
For the cannula implantation surgery, after the social interaction test, which was performed the day after the last social defeat stress exposure, mice were anesthetized with isoflurane (induction with 3% and maintenance with 2% through a mask) using MK-A110 (MUROMACHI KIKAI CO., LTD. Japan), and the heads were fixed in a stereotaxic apparatus (Model 942, David Kopf Instruments, USA). Bilateral guide cannulae (RWD-62064, length: 6.0 mm, RWD Life Science, China) with a dummy cannula (RWD-62164, length: 6.2 mm, RWD Life Science) was implanted into the VTA (AP: −3.28 mm from the bregma, ML: ±0.5 mm from the bregma, DV: −4.25 ± 1.5 mm from the dura). The cannula was secured with dental cement (56849, 3M Company, USA).
For the intra-VTA infusion, raclopride (final concentration: 0.1 mM) was dissolved in aCSF (3525, R&D Systems, USA). More than 4 days after surgery, just before intraperitoneal administration of ketamine, raclopride was delivered into the VTA using an injection cannula (RWD-62264, length: 6.2 mm, RWD Life Science) connected with polyethylene tubing (427411, BD Intramedic, USA). The drugs were infused (0.5 μl/hemisphere) at a rate of 0.1 μl/min using Hamilton syringes (84853, Hamilton, USA) and a syringe pump (LEGATO 180, KD Scientific, USA). The cannula placement was confirmed by histological analysis of coronal brain slices. Mice with inaccurate cannula placements were excluded from the study.
Locomotor activity test
The mice were placed in an acrylic cylinder (19 cm diameter), and the horizontal locomotor activity was measured for 180 min (interval: 5 min) by automatic actigraphy (SCANET MV-40; Melquest, Japan). Ketamine was administrated 90 min after the start of the session. Aripiprazole and brexpiprazole were injected 15 min before ketamine administration. The locomotor activity of each mouse was normalized using locomotor activity before administration. Outliers are removed using Smirnov-Grubbs test.
Forced swim test
Mice were placed in a plastic cylinder (50 cm height × 8 cm diameter) filled with water at a temperature of 25 ± 1 °C and a depth of 35 cm. The duration of immobility was measured during the first 6 min. The immobility time was measured when no additional activity was observed other than that required to keep its head above the water. The analyses were conducted under a blinded condition about the group allocation.
SIT
The SIT was performed as previously reported [36,37,38]. B6 mice were individually placed in an open arena (42 cm width × 42 cm depth × 42 cm height) with an empty wire-mesh enclosure (10 cm width × 6.5 cm depth × 42 cm height) and given 3 min to explore the arena (empty session). Immediately afterwards, the mouse was removed from the arena. The mouse was placed again within the same open arena, but this time with the wire cage containing a novel ICR mouse, for 3 min (target session). Time spent in the area surrounding the wire cage (interaction zone, 42 × 14 cm) was automatically measured with an ANY-maze tracking system (Stoelting, USA). SI ratio was calculated by dividing the interaction zone time in target session by the interaction zone time in empty session.
Brain slice preparation for histology
For histological analyses, 105 min after drug administration, the mice were anesthetized with isoflurane and transcardially perfused with PBS followed by 4% paraformaldehyde (PFA). The brains were then removed and post-fixed with 4% PFA overnight at 4 °C. Thereafter, the brains were placed in 30% sucrose in PBS for >24 h, and then frozen in OCT compound (4583, Sakura Finetek, Japan). The brains were coronally sectioned at a thickness of 20 μm with a cryostat (CM1860 UV, Leica). The sections were washed with PBS 3 × 10 min prior to immunohistochemistry and in situ hybridization/immunohistochemistry staining procedures.
Immunohistochemistry
All steps were performed at room temperature (RT), unless otherwise indicated. The sections were blocked with 3% bovine serum albumin in TBS for 1 h. Then, the sections were immunostained with rabbit anti-c-Fos antibody (1:1000; #2250, Cell Signaling Technology, USA) and mouse anti-rat tyrosine hydroxylase antibody (1:1000; 556311, BD Pharmingen, USA) diluted in 0.5% Triton X-100 in TBS at 4 °C overnight. The sections were rinsed with TBS 3 × 10 min and incubated with Alexa Fluor 488-conjugated goat anti-rabbit IgG secondary antibody (1:1000; #A11034, Thermo Fisher Scientific, USA) and Alexa Fluor 594-conjugated goat anti-mouse IgG secondary antibody (1:1000; #A11034, Thermo Fisher Scientific) diluted in TBS for >5 h at 4 °C.
Combined in situ hybridization/immunohistochemistry
All steps were performed at RT, unless otherwise indicated. The sections were incubated with a rabbit anti-c-Fos antibody (1:1000; #2250, Cell Signaling Technology) and/or a mouse anti-rat tyrosine hydroxylase antibody (1:1000; #556311, BD Pharmingen) 1–2 days before in situ hybridization. The in situ hybridization for D2R was performed using the RNAscope Fluorescent Multiplex Kit V2 (Advanced Cell Diagnostics, USA) as per the manufacturer’s instructions, with minor modifications. Briefly, the sections were fixed in 4% PFA for 10–30 min and subsequently dehydrated for 10–30 min with Protease Plus at 40 °C. After washing with PBS, the sections were incubated with Mm-Drd2 probe-C1 (406501) for 50–120 min at 40 °C. After washing, the signal for the C1 probe was amplified with horseradish peroxidase (HRP)-C1 for 10–15 min, followed by Opal620 (1:1000; OP-001004, Akoya Biosciences, USA) for 20–30 min at 40 °C. Then, the C1 channel was blocked with an HRP blocker for 10–15 min at 40 °C. For the detection of the TH signal, the sections were incubated with HRP-conjugated goat anti-mouse IgG (1:1000; #A16078, Thermo Fisher Scientific) for 30 min followed by amplification with Opal690 (1:1000; OP-001006, Akoya Biosciences) for 15 min. For the detection of the Fos signal, the sections were incubated with HRP-conjugated goat anti-rabbit IgG (1:1000; A16110, Thermo Fisher Scientific) for 30–60 min followed by amplification with Opal520 (1:1000; #FP1487A, PerkinElmer, USA) for 15 min at RT or 20 min at 40 °C. For the incubation at 40 °C, a HybEZ oven (Advanced Cell Diagnostics) was used.
Imaging and image analysis
After immunohistochemistry and combined in situ hybridization/immunohistochemistry, the sections were rinsed with TBS or PBS 3 × 10 min and mounted with FluorSave mounting medium (345789, Merck Millipore, USA). For multiple staining, the sections were counterstained with DAPI (340–07971, Wako Pure Chemical Industries) before mounting. Images were obtained on a fluorescence microscope (BZ-X710, KEYENCE, Japan) and merged using the BZ-X Analyzer software (KEYENCE). Cell counting was performed with ImageJ software (National Institutes of Health) and a deep learning-based model for cell counting [40].
Hierarchical clustering and network analysis using Fos data
Fos+ cell density was normalized by the mean value of each brain region of the vehicle-treated groups. Unsupervised hierarchical clustering was performed using Ward’s hierarchical agglomerative clustering method on the Euclidean distances of the row-wise z-scores of Fos+ cell densities in the brain regions using Seaborn library in Python. Regions with the smallest distances, indicating high similarity, were grouped iteratively. A correlation matrix was generated by computing Pearson correlation coefficients of Fos densities between pairs of the brain regions. A weighted undirected network was constructed with correlations with Pearson’s |r|> 0.5. The nodes in the network represent brain regions, and the correlations that survived thresholding were shown as connections. Network construction and eigenvector centrality was calculated using NetworkX package in Python. Eigenvector centrality measures a node’s influence by the centrality of its connections, valuing nodes linked to well-connected neighbors. High eigenvector centrality indicates significant influence through these high-quality connections within the network.
Statistical analysis
All data are expressed as the mean ± SEM. Before group comparison, the normality of distribution was assessed using the Shapiro-Wilks normality test. For single comparisons, paired t and Wilcoxon matched-pairs signed rank tests were used for normal and non-normal distribution data, respectively. For multiple comparisons, Dunnett’s multiple comparisons and Dunn’s multiple comparisons tests were used for normal and non-normal distribution data, respectively. For multiple comparisons with two independent variables, two-way repeated-measures ANOVA was used. Post-hoc test was used depending on the number of groups (two groups: Šídák’s multiple comparisons test, three groups: Fisher’s LSD test, >3 groups: Dunnett’s multiple comparisons test). Type of statistical method and sample size of each experiment are described in figure legends. p < 0.05 was considered statistically significant. All statistical analyses were carried out using GraphPad Prism 6 or 10 (GraphPad Software, USA).
Clinical study
Study Design
The clinical study was a single-arm, double-blinded clinical study of sequential treatments including ketamine infusion and oral administrations of aripiprazole and brexpiprazole in depressed patients. The subjects and the examiners were blinded about the medications and the treatment schedule. Assuming that the study drug has the efficacy to completely suppress ketamine-induced dissociation symptoms, and assuming a one-sided significance level of 0.05, the number of patients required to detect a significantly lower change in CADSS score with the study drug than without the study drug (treatment with ketamine alone) by Wilcoxon test with a probability of more than 80% was calculated. Based on the report by Wan LB et al. [41], the required number of patients was calculated to be 10 when the difference between the CADSS score before and after ketamine administration was 9.8, the combined standard deviation before and after administration was 10, and the effect size was 0.9. Therefore, the target sample size was set 10, and the actual number of recruited subjects was 9.
Subjects
Participants were required to be at least 18 years of age, diagnosed with depression according to DSM-5 criteria, and have persistent symptoms for at least 4 weeks, with MADRS scores of 17 or higher and BDI-II scores of 14 or higher. They must have had an inadequate response to at least one adequate dose of antidepressant for at least 1 month or be intolerant to at least two antidepressants for a sustained episode of at least 1 month. Written consent to participate was required.
Exclusion criteria: Exclusions were based on serious physical illness, a history of ketamine non-responsiveness, concomitant or previous psychiatric or neurological disorders, or strong suicidal ideation.
Restricted medication
Subjects were instructed to cease the consumption of foods and drugs strongly affecting CYP3A4 28 days before the first ketamine dose until the study concluded. The subjects were required to discontinue antipsychotics at least for a period five times the blood half-life. No new psychiatric treatment was introduced, except for treatment received up to 14 days before the study’s initiation.
Schedule
As shown in Fig. 4a, subjects received an intravenous dose of 0.5 mg/kg ketamine over 40 min on days 1, 5, 8, 12 and 15 of a 16-day period. Four hours prior to ketamine administration, subjects received placebo (day 1), aripiprazole 3 mg (day 5), aripiprazole 12 mg (day 8), placebo (day 12; washout), and brexpiprazole 2 mg (day 15) orally in a blinded manner.
Psychological assessments were conducted three times for each ketamine dose, timed before taking the study drug, 40 min after ketamine administration, and 20–28 h after the start of ketamine administration. A blood sample was taken 40 min after ketamine administration, and plasma concentrations of ketamine, aripiprazole and brexpiprazole were measured. Aripiprazole blood levels of 20 ng/ml are used as the cut-off. This threshold enables differentiation between aripiprazole 12 mg and 3 mg doses, as the highest blood concentration after a 3 mg dose is approximately 15 ng.
General biochemical tests, ECG assessments and vital sign checks were performed on the day of each ketamine dose, prior to administration, to observe adverse events.
Psychological evaluation
Blinded psychologists assessed the primary endpoint, the Clinician-Administered Dissociative States Scale (CADSS), as well as secondary endpoints, including MADRS, the Brief Psychiatric Symptom Rating Scale/Positive Symptom Subscale (BPRS-P), the Visual Assessment Scale (VAS), the Sedation Rating Scale (MOAA/S) and the Colombian-Suicide Severity Rating Scale (C-SSRS) for safety. When evaluating the primary endpoint (dissociative symptoms), a score of at least 4 and an increase of at least 1 in the CADSS score were used as thresholds for Positive/Negative assessment, similar to the criteria in the package insert for esketamine (SPRAVATO).
Statistical analysis
All data are expressed as the mean ± SEM. Before group comparison, the normality of distribution was assessed using the Shapiro-Wilks normality test. For single comparisons, one-tailed paired t and one-tailed Wilcoxon matched-pairs signed rank tests were used for normal and non-normal distribution data, respectively. For multiple comparisons with one independent variable, repeated-measures ANOVA and Friedman ANOVA test were used for normal and non-normal distribution data, respectively. Mixed-effects model was applied to data with missing values. Dunnett’s multiple comparisons and Dunn’s multiple comparisons tests were used as post-hoc tests for normal and non-normal distribution data, respectively. For contingency table comparison, Fisher’s exact test was used. Type of statistical method and sample size of each experiment are described in figure legends. p < 0.05 was considered statistically significant. All statistical analyses were carried out using GraphPad Prism 6 or 10 (GraphPad Software, USA).



Responses