A three-step strategy for the conversion of pyridines into benzonitriles
Main
Bioisosteric replacement is a pivotal strategy extensively used during critical phases of drug discovery campaigns1. At the lead optimization stage, it is often used in library generation for both hit expansion and structure–activity relationship (SAR) studies. Additionally, it is an effective method to circumvent patent protection, something generally referred to as ‘scaffold hopping’2.
Although the bioisosteric replacement of specific peripheral functional groups can be performed in a modular and programmable fashion (for example, via amidation, cross-coupling or C–H activation), the bioisosteric replacement of a core ring structure within a bioactive molecule often necessitates a ground-up redesign of the entire synthetic sequence. This endeavour is necessarily time consuming and costly3. Consequently, reactions enabling modular late-stage ‘ring replacement’ allow for rapid diversification in retrosynthetically unconventional manners. Such processes are in high demand yet remain rare due to the considerable synthetic challenges they pose4,5,6,7,8,9,10,11,12,13,14,15,16.
A highly sought-after ring replacement in medicinal chemistry involves substituting benzene with pyridine because the incorporation of a nitrogen atom in the arene can often boost biological potency. This phenomenon, frequently observed and now termed the ‘necessary nitrogen effect’, is mainly due to the inherent metabolic stability imparted by the nitrogen atom (pyridine is more difficult to oxidize than benzene) and the potential for hydrogen-bond interactions through its lone pair of electrons17,18,19,20,21.
However, given the ubiquity of pyridine-containing bioactive material22, the opposite direction of ring replacement, namely, pyridine to benzene, can also provide many opportunities for SAR studies (Fig. 1a). Among the various types of benzenoid systems, benzonitriles hold a privileged position. A nitrile group can effectively polarize the aromatic ring similarly to pyridine, thereby enhancing metabolic stability in most cases23,24,25,26,27. In practice, two main types of pyridine-to-benzonitrile replacements have demonstrated strong potential and consequent applicability in medicinal chemistry. First, 2-substituted benzonitriles have been identified as effective bioisosteres for 4-substituted pyridines23,28,29,30,31,32,33 (Fig. 1b). This is particularly important given that the 4-substituted pyridine motif appears in nearly one-third of all US Food and Drug Administration (FDA)-approved drugs featuring this azine core22. Furthermore, another commonly used replacement involves substituting the pyridine nitrogen atom with a ‘C–CN’ unit because the nitrile group can mimic the pyridine hydrogen-bond acceptor ability34 (Fig. 1c). This approach has been particularly useful when one bridging H2O molecule is involved in the binding of a bioactive pyridine with its biological target27. This bioisosteric replacement can effectively displace the ‘unhappy water’ from the interaction site, thereby reducing the entropy of binding35,36. Such a strategy has often been pivotal in the development of commercial drugs such as neratinib and bosutinib from Pfizer or other highly active molecules such as a p38 inhibitor under development by Bristol-Myers Squibb27,37,38,39.

a, The bioisosteric replacement of pyridines with benzonitriles is often used in medicinal chemistry studies23,28. b, 2-Substituted benzonitriles are powerful bioisosteric replacements for 4-substituted pyridines29,30,31,32,33. c, The replacement of the pyridine nitrogen atom with a ‘C–CN’ unit can improve biological activity by displacement of ‘unhappy water’ molecules27,35,36. d, This work provides a three-step method for the conversion of 4-substituted pyridines into 2-substituted benzonitriles. EWG, electron-withdrawing group; IC50, half-maximal inhibitory concentration; [ox], oxidation.
Overall, designing a synthetic strategy for pyridine-to-benzonitrile replacement might be a useful tool for SAR studies and scaffold hopping purposes. Here we present a photochemical strategy that converts 4-substituted pyridines into 2-substitute benzonitriles in just three steps (Fig. 1d). The method requires initial pyridine N-oxidation, for photochemical deconstruction into a nitrile-containing linear intermediate. This species is then rearomatized into the corresponding 2-substituted benzonitrile via Diels–Alder cycloaddition with various dienophiles. This process leverages the externalization of the pyridine nitrogen atom into the nitrile functionality, thereby bypassing the need for toxic cyanide sources typically used in transition-metal-mediated cyanation strategies40.
Results and discussion
Design plan
The design of our three-step process is based on a strategic photochemical pyridine deconstruction step to form an acyclic-nitrile-containing intermediate (Fig. 2a). For this step to occur, initial pyridine activation is required by N-oxidation generally with meta-chloroperoxybenzoic acid (mCPBA) (step a, I → II). Pleasingly, many pyridine-N-oxides are also commercially available, thus reducing the pyridine-to-benzonitrile replacement to effectively two steps.
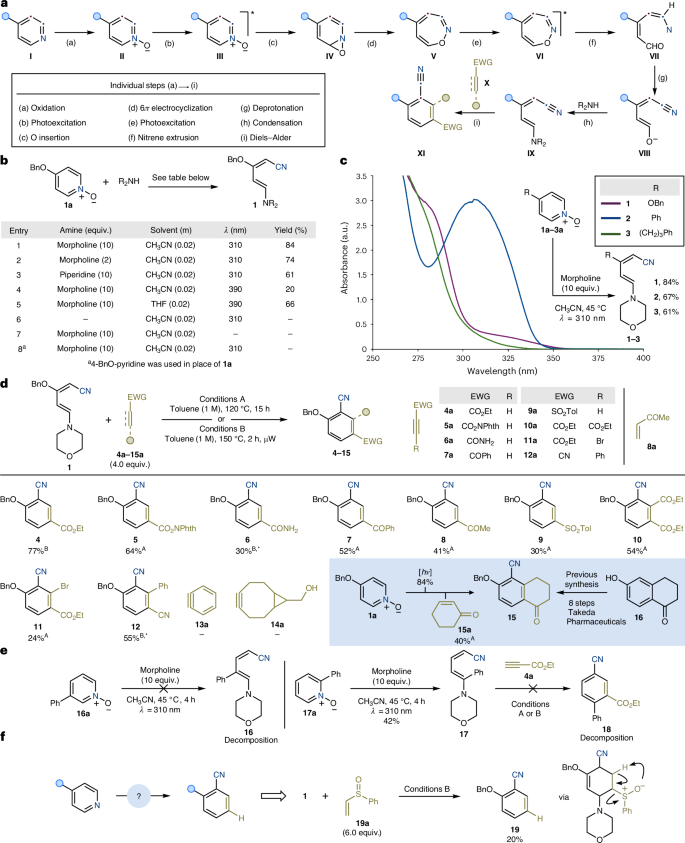
a, Mechanistic pathway for the three-step conversion of 4-substituted pyridines into 2-substituted benzonitriles. b, Reaction development for the photochemical conversion of 1a into 1. c, Ultraviolet–visible absorption spectroscopy studies. d, Diels–Alder cycloaddition scope and applications. e, Current limitations. f, Use of (vinylsulfinyl)benzene as dienophile enables access to 2-substitued benzonitriles without substituents at C5. Superscripts A and B indicate reactions performed using conditions A and B, respectively. *See Supplementary Information, section 11.2, for further details. Bn, benzyl; λ, irradiation wavelength; Phth, phthalimide
In the key photochemical deconstruction event, one pyridine endocyclic C–N unit is transformed into a nitrile group, while creating a dienic structure (IX) capable of participating in ring-constructing reactions, such as Diels–Alder cycloaddition, with a second synthon (IX + X → XI)41,42,43,44,45,46. For the deconstruction step, we drew inspiration from the pioneering work of Buchardt, Lohse and Albini showing that 4-substituted pyridine-N-oxides, upon irradiation with a mercury lamp in the presence of an amine, undergo a photochemical ring opening sequence producing an aminopentadienenitrile intermediate. However, this reactivity proved low yielding, giving the desired product alongside 2-formyl pyrrole derivatives and tarring under the stated conditions47,48,49,50,51,52,53. Mechanistically, it has been postulated that, upon irradiation, II populates its S1 state that has π,π* character (step b). This excited state species would then initiate an oxygen-atom insertion to provide a transient oxaziridine (step c, IV)54, evolving by electrocyclic 6π ring opening into a 1,2-oxazipine V (step d)55. At this point, a second photon absorption event would deliver the S1–VI (step e) from where a triplet nitrene can be extruded (step f, VI → VII)56. Exploitation of the intrinsic acidity of the α-nitrene C(sp2)–H bond in VII allows for deprotonation to generate the nitrile functionality in VIII (step g)57,58,59,60. The resulting enolatepentadienenitrile VIII can then condense with an amine to furnish the desired aminopentadienenitrile IX (step h).
For the third and final step of our strategy, we postulated that IX would be sufficiently electron-rich to undergo [4πs + 2πs] Diels–Alder cycloaddition followed by concomitant aromatization with a suitably electron-deficient dienophile X (step i).
Overall, the successful implementation of this synthetic design would generate a benzonitrile species XI in which the nitrogen atom has been externalized and converted into a nitrile functionality, while the pyridine substituent at the 4-position has been translated, in a programmable and regioselective manner, to its 2-position. Moreover, the introduction of a difunctionalized dienophile at the cycloaddition stage can potentially enable simple access to 1,2,3,4-tetrasubstituted aromatics, which are challenging synthetic derivatives due to their sterically congested cores61.
Reaction development and understanding
We proceeded with the optimization of the key pyridine deconstructive step by using the commercial 4-BnO-pyridine-N-oxide 1a (Fig. 2b). Pleasingly, irradiation (λ = 310 nm) of 1a and morpholine (10 equiv.) in CH3CN solvent at room temperature for 15 h provided diene 1 in high yield (entry 1). The use of lower amounts of morpholine (entry 2) or piperidine as the amine (entry 3) successfully delivered 1 albeit in slightly diminished yields.
Ultraviolet–visible absorption spectroscopy analysis on 1a revealed high absorption in the 280–330 nm region with a tail reaching the near-visible region (Fig. 2c). However, utilization of purple light-emitting diodes (λ = 390 nm) resulted in low conversion in the deconstructive process (entry 4). Interestingly, we noticed a considerable solvochromic effect, and in tetrahydrofuran, the absorption profile of 1a showed a notable bathochromic (red) shift, which in turn enabled the use of lower-energy light sources for the conversion into 1 (entry 5) (Supplementary Information). Control experiments in the absence of either morpholine (entry 6) or light irradiation (entry 7) or using 4-BnO-pyridine in place of the N-oxide 1a (entry 8) resulted in no product formation.
Because this photochemical event is intrinsically reliant on the absorption profile of the various pyridine-N-oxide reagents, we quickly evaluated other derivatives featuring different electronic perturbations of the azine ring. In particular, 4-Ph and 4-alkyl derivatives 2a and 3a display a markedly lower absorption profile, but using λ = 310 nm we obtained the desired dienes 2 and 3 in good yields (Fig. 2c).
With a strategy for pyridine-N-oxide deconstruction in place, we then focused our attention on the following rearomatization step via Diels–Alder cycloaddition (Fig. 2d). This was initially performed on isolated diene 1, which was routinely prepared on up to 5-mmol scales using our aforementioned photochemical ring-opening methodology. Although the supplementary material describes all optimization efforts using ethyl propiolate 4a (Supplementary Information, section 4, and Supplementary Tables 10–14), we identified two simple thermal conditions using toluene as the solvent. In general, reactive dienophiles could be engaged at 120 °C (conditions A), whereas less-reactive ones required more forcing conditions, which were met with the use of a standard microwave apparatus (conditions B). Under these conditions, we successfully accessed benzonitriles 4–15 featuring meta-ester (4 and 5), primary amide (6), ketone (7 and 8) and sulfone (9) functionalities. It is interesting to note that the cycloaddition does not require the use of an alkyne dienophile as 8 was obtained using Me-vinyl-ketone 8a. In this case, we propose that the cycloadduct intermediate undergoes morpholine elimination followed by aromatization by hydride transfer to the enone (Supplementary Information, section 12, and Supplementary Fig. 15). The formation of 5 is also synthetically noteworthy as the redox-active ester functionality can now be used in several radical-based cross-coupling manifolds for further functionalization62,63,64. Disubstituted alkynes were screened next and enabled access to tetrasubstituted aromatics with additional ester (10), bromine (11) and aryl (12) substituents. In terms of limitations, we did not succeed in engaging benzyne 13a and the ‘click chemistry’ alkyne 14a in the process. Preliminary computational studies indicated that the lack of a functionality on the alkyne of 14a inhibits reactivity, while the finding for 13a is probably a consequence of the high reactivity of benzyne (Supplementary Information, section 12, and Supplementary Fig. 41).The retrosynthetic opportunity that this approach might provide for the preparation of functional materials was demonstrated by the synthesis of tetralone 15. This species has been recently patented by Takeda Pharmaceuticals as an intermediate in the synthesis of an API targeting the β-adrenoceptor65,66. Interestingly, its preparation was achieved using an eight-step synthetic route starting from phenol 16. The unusual tactic introduced by our methodology simplified its preparation, shortening it to just two steps using commercial 1a and 15a.
A key observation in terms of limitations was that it could not be extended to 3- and 2-substituted pyridine-N-oxides such as 16a and 17a (Fig. 2e). In the case of 16a, photochemical ring opening resulted in a complex mixture of products, possibly owing to the instability of its corresponding nitrene intermediate. By contrast, 17a underwent efficient ring opening; however, the following cycloaddition with diene 17 could not be achieved because it led to extensive decomposition.
At the moment, our strategy enables the conversion of 4-substituted pyridines into 2-substituted benzonitriles with an additional electron-withdrawing substituent placed at C5. Although this group is necessary to favour the Diels–Alder step, it would be synthetically valuable to identify conditions to access C5-unsubstituted derivatives (Fig. 2f). This proposal would require a challenging cycloaddition step with gaseous acetylene. Hence, we decided to identify a potential synthon acting effectively as an acetylene surrogate. Specifically, we were pleased to identify that the use of (vinylsulfinyl)benzene 19a as the dienophile led to the direct formation of 19 in 20% yield. We believe the initial sulfoxide-containing cycloadduct undergoes a tandem Ei-type elimination followed by aromatization by morpholine elimination67,68.
The final cycloaddition event has not been studied before, and therefore we initiated a computational study with 1 and methyl propiolate (I) as the model partners (Fig. 3a). After a detailed conformational search of possible intermediates and transition states (Supplementary Information, section 12, and Supplementary Figs. 25 and 26) we found that the lowest-energy conformation of 1 has a s-trans arrangement. However, the lowest-energy transition states located for cycloaddition with I proceed through an s-cis conformation (ΔG‡ = 38.1 kcal mol–1), although the penalty of 1 adopting this reactive conformation is ΔG° = 2.7 kcal mol–1. Considering the whole ensemble of feasible transition state structures of 1 reacting with A, it is noted that all occurred with a key r(C3–C4) bond distance >2.92 Å and a much shorter r(C1–C2) averaging 1.95 Å. This suggests the process occurs via an energetically concerted, highly asynchronous Diels–Alder process to give C that can eliminate morpholine to D or a more complex mechanism potentially through a stepwise process, potentially via intermediate B.
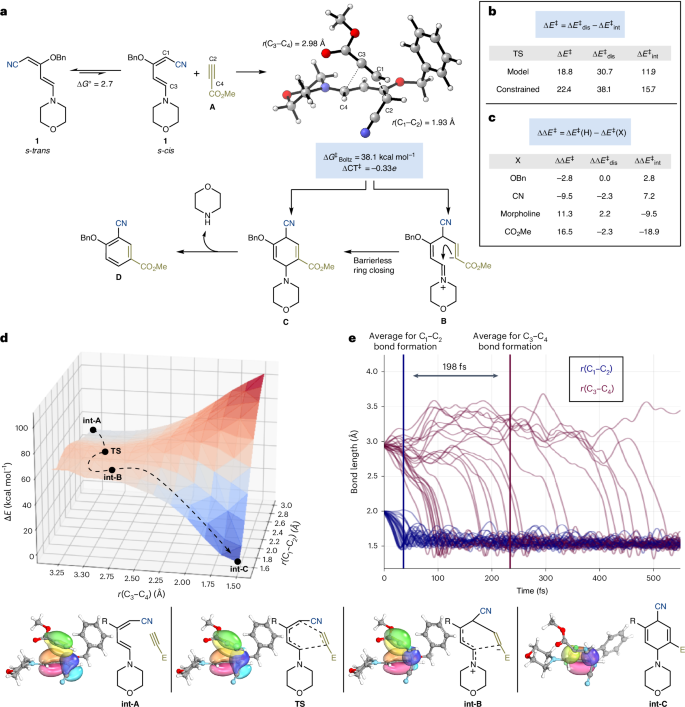
a, Proposed mechanism for formal cycloaddition. b, Distortion-interaction analysis conducted on an example transition state and a species with constrained bond lengths. c, Effects of substituents on the example transition state by their removal and capping with a hydrogen atom at the transition state geometry. Energy values in b,c are displayed in kcal mol−1. d, More O’Ferrall–Jencks plot for a truncated system of the model transition state (CPCM(Toluene)-M06-2X/ma-def2-SVP), with an idealized reaction path. e, Analysis of Born–Oppenheimer molecular dynamics (CPCM(Toluene)-M06-2X/ma-def2-SVP), initiated from the truncated transition state, examining the time taken for the key bond formation. General method: SMD(Toluene)-M06-2X/ma-def2-QZVPP/CPCM(Toluene)-M06-2X/ma-def2-SVP. Orbitals: SMD(Toluene)-M06-2X/def2-TZVP/CPCM(Toluene)-M06-2X/ma-def2-SVP. Molecular dynamics: CPCM(Toluene)-M06-2X/ma-def2-SVP. E, CO2Me; R, OBn; TS, transition state; for 3D structures, grey atoms are carbon, white hydrogen, red oxygen and blue nitrogen.
To understand the underlying physical contribution, we applied a distortion-interaction analysis to a model transition state primed for a concerted addition 69,70,71,72,73, which revealed that for this system the distortion energy is over twice the interaction energy (ΔE‡dis = 30.7 kcal mol–1 and ΔE‡int = 11.9 kcal mol–1) (Fig. 3b). However, if the transition state is constrained to a more synchronous Diels–Alder type reactivity (r(C1–C2) = 2.10 Å and r(C3–C4) = 2.35 Å) a slight increase in interaction energy is observed (ΔE‡int = 15.7 kcal mol–1) but this does not compensate for the additional distortion (ΔE‡dis = 38.1 kcal mol–1). These findings further suggest that a synchronous Diels–Alder reaction does not provide sufficient energetic gains because it leads to a notable increase in strain.
The exemplar transition state was further studied with an analysis inspired by the work of Wheeler74,75,76,77, in which the substituents were individually removed and capped with a hydrogen atom to discern their specific effects (Fig. 3c). These data point towards the -CN and -OBn groups contributing to the transition state through destabilizing interactions (ΔΔE‡ < 0 and ΔΔE‡int > 0) with minimal additional strain. This effect is offset by the morpholine and carboxylate groups, which now predominantly act to stabilize the transition state through the interaction term (ΔΔE‡ > 0 and ΔΔE‡int < 0). Overall, this analysis suggests the key strain energy associated with the transition state is intimately associated to its distorted six-membered-ring core with minimum impact from the various substituents.
Further analysis of the transition state revealed the presence of charge-transfer from the diene to the alkyne (ΔCT‡ = –0.33e). This can potentially be explained through the formation of a zwitterionic intermediate B undergoing cyclization to C. Our analysis suggested this process to be barrierless, hence potentially involving an ‘entropic dynamic intermediate’78,79. To provide insights into this, we calculated intrinsic bonding orbitals80,81 along the intrinsic reaction coordinates, which provide an intuitive way to understand the flow of electrons throughout the reaction, depicted on an ideal path along the More O’Ferrall–Jencks plot for a model system82 (Fig. 3d). The intrinsic bonding orbital at the beginning of the reaction clearly indicates three π-bonds delocalized by their neighbouring environment (int-A); on progressing to the transition state two π-bonds spatially change with one converting to a σ-bond and the other a π-lone pair (TS). This is again in stark contrast to a standard Diels–Alder reactivity in which the other π-bond is involved to form a second σ-bond. This finding further reinforces the likelihood that even when the alkyne is aligned with the diene, the cycloaddition progresses through a stepwise zwitterionic entropic intermediate (TS → int-B → int-C). Therefore, to determine whether the aligned system truly went through a dynamically concerted asynchronous Diels–Alder process or a dynamically stepwise process, Born–Oppenheimer molecular dynamics were applied to a model system at a suitable transition state geometry83,84,85,86 (Fig. 3e). After random initiation, the productive runs were shown to form r(C1–C2) < 1.6 Å on average in 37 fs; by contrast, the r(C3–C4) bond occurred 198 fs later and one trajectory did not even form a bond after >1,000 fs. The smallest time gap between the two bond-forming processes found was 52 fs, which is at the upper timescale of a C–C bond vibration (30–60 fs)87, hence we propose that the reaction occurs through a dynamic stepwise mechanism forming an entropic zwitterionic intermediate before cyclizing to the productive Diels–Alder adduct85.
Scope of the process
Having identified suitable conditions for the ring opening of pyridine-N-oxides and subsequent benzonitrile construction, we sought to combine both reactions into a one-pot process, thereby avoiding the isolation of any synthetic intermediate. This was achieved through directly telescoping both sets of reactions utilizing only evaporation of volatiles after the photochemical process to facilitate the switch in solvent and allow for the removal of the remaining morpholine (Fig. 4a).
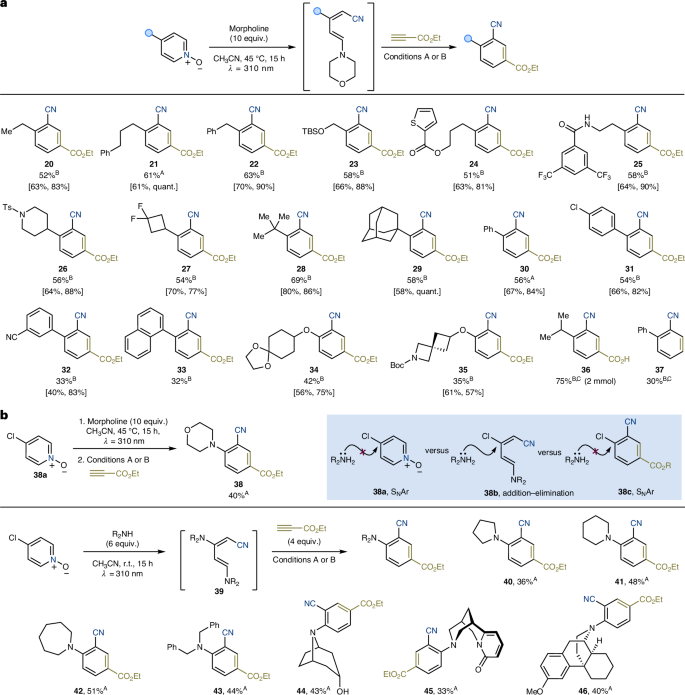
a, Pyridine scope. b, Tandem pyridine deconstruction–amination–Diels–Alder reactivity to give ortho-aminated benzonitriles. [Yield of step 1 (ring opening), yield of step 2 (cycloaddition)]. Superscripts A and B indicate reactions performed using conditions A and B (see Fig. 2 for details); for details of superscript C, see Supplementary Information. TBS, tert-butyldimethylsilyl; Ts, p-toluenesulfonyl; Boc, tert-butoxycarbonyl.
We started scope exploration by first evaluating 4-alkyl pyridines. This enabled the use of primary groups (20–25) including derivatives with positions that are enthalpically (22, benzylic) or polarity (24, α-O) activated towards hydrogen-atom transfer88,89. This evaluation also demonstrated tolerance of silyl-protected alcohol (23), ester (24) and amide (24) functionalities. The reactivity was also extended to substrates containing secondary and tertiary alkyl substituents, such as a 4-N-Ts-piperidine (26) and a gem-difluoro-cyclobutane (27), and bulky t-Bu (28) and adamantyl (29) groups. It is interesting to note that many of these 4-alkyl-pyridine-N-oxides are accessed by straightforward Minisci reactivity90,91,92,93. This means that radical chemistry can be used as a stepping stone to functionalize in a selective manner the pyridine starting material for subsequent translation into the benzonitrile product. This offers valuable complementarity to the preparation of derivatives that would otherwise require C(sp2)–C(sp3) cross-couplings with transition metals94,95.
4-Aryl groups were explored next, and the two-step one-pot cascade allowed for the synthesis of Ph (30), 4-Cl-C6H4 (31), 3-CN-C6H4 (32) and 1-naphthyl (33) containing biaryls.
Ortho-substituted benzonitrile ethers were prepared starting from 4-hydroxy-pyridine via a Mitsunobu reaction with secondary alcohols. Subsequent photochemical deconstruction and thermal cycloaddition gave 34 and 35 in good yield.
The scalability of the overall process was explored using 4-i-Pr-pyridine-N-oxide, which was converted in three telescoped steps (deconstruction, cycloaddition, ester hydrolysis) into the benzonitrile 36 on a 2-mmol scale in overall 75% yield. Furthermore, the use of 19a could be adapted in the telescoped approach to give 37 in overall 30% yield.
An interesting outcome was obtained while extending this chemistry to 4-Cl-pyridine-N-oxide 38a, which under the telescoped conditions delivered the 2-aminated benzonitrile 38 (Fig. 4b). Because 38a is a commercial building block often used in nucleophilic aromatic substitution (SNAr) chemistry, we initially reasoned that 38a was undergoing in situ amination before ring opening en route to 38. Alternatively, we hypothesized that morpholine expelled upon aromatization after the cycloaddition step might have resulted in a tandem Diels–Alder–SNAr sequence. However, control experiments revealed that no SNAr took place either with 38a (45 °C) or 38c (120 °C). This outcome is not surprising because these conditions are effectively base-free, which would limit the nucleophilic properties of the amine. Furthermore, several literature methods for SNAr aminations were attempted 38c and in all cases, the yield of the desired aminated benzonitrile did not exceed 14% (Supplementary Information, section 12, and Supplementary Table 17, entries 3–4). These observations suggested that ring opening occurred to generate chlorine-substituted aminopentadienenitrile 38b, which undergoes addition–elimination with the amine to give 39 before Diels–Alder reactivity.
Recognizing the potential of this reactivity for tandem pyridine-to-benzonitrile conversion and ortho-amination, we explored the compatibility of the telescoped process with various amine coupling partners. Pleasingly, pyrrolidine (40), piperidine (41), azepane (42) and Bn2NH (43) were tolerated, giving their constituent 2-aminobenzonitriles 40–43 in moderate to good yield. The ability of the strategy to deliver complex materials was further demonstrated by the arylation of alkaloid nortropine (44), the smoking cessant cytisine (45) and nor-dextromethorphan (46).
We believe this strategy for pyridine-to-benzonitrile ring replacement can have two main applications.
Considering the large number of pyridine-containing drugs, it can find use as a late-stage ‘ring-replacer’ to access new types of derivatives without the need for de novo individual synthesis, which is the current-state-of-the-art (Fig. 5a). This concept was showcased with the three-step modification of the anticancer opaganib (47a), the HBB inhibitor 48a and the pupil dilator tropicamide (49a). These bioactive materials feature a 4-substituted pyridine core and underwent N-oxidation followed by photochemical deconstruction and thermal reconstruction with ethyl propiolate in overall good yields (47–52). In the case of 49a, the photochemical step was run on a 0.3-mmol scale, which enabled parallel preparation of other derivatives (49, 50, 51) by changing the nature of the dienophile. We hope this can provide a powerful opportunity for library screening purposes by exploiting, in a modular manner, already established bioisosterism principles.
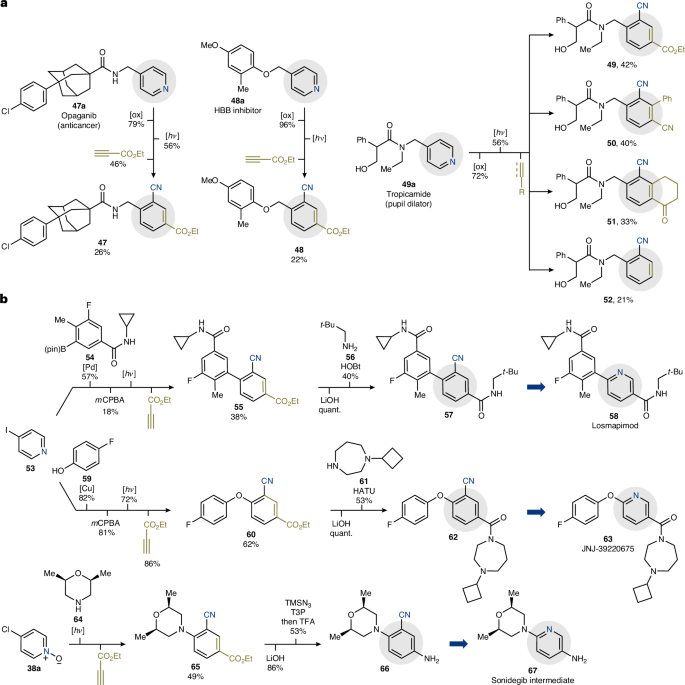
a, Use of the methodology for the late-stage pyridine-to-benzonitrile ring replacement of drugs. b, Use of the method for the preparation of ‘unhappy water’ analogues of pyridine drugs. B(pin), pinacol boronic ester; HATU, hexafluorophosphate azabenzotriazole tetramethyl uronium; T3P, propylphosphonic anhydride; TMSN3, trimethylsilyl azide; TFA, trifluoroacetic acid. See Supplementary Information, section 11.5, for details of reaction conditions.
Furthermore, the alternative tactic for aromatic construction provided by this method can streamline the preparation of functionalized benzonitriles from unusual pyridine starting materials (Fig. 5b). We were keen to showcase this unique retrosynthetic opportunity and decided to prepare the ‘unhappy water’ benzonitrile analogues of pyridine-based losmapimod (58), JNJ-39220675 (63) and the key aniline fragment of sonidegib (67). The syntheses of 57 and 62 started from 4-iodopyridine 53 that underwent either Suzuki–Miyaura cross-coupling with 54 (three steps) or Ulmann coupling with phenol 59. These steps provided access to 4-functionalized pyridines that were subjected to the three-step procedure for benzonitrile construction. Pleasingly, both derivatives were converted into 55 and 60, respectively, in good yields. At this point, ester hydrolysis followed by amidation with either 56 or 61 provided the ‘unhappy water’ analogues 57 and 62 in five steps. The preparation of the sonidegib intermediate 67, harnessed the tandem amination discussed above (Fig. 4b). Hence, commercial 4-chloropyridine-N-oxide 38a was directly converted into the morpholine-containing derivative 65 in good yield. Subsequent hydrolysis and Curtius rearrangement led to aniline 66 in four steps. This highlights the versatility of the ester substituent which can be manipulated as a synthetic handle for further downstream functionalization in the synthesis of complex aromatic targets.
Conclusions
We have presented a method for the direct conversion of pyridines into benzonitriles. This strategy harnesses pyridine N-oxidation as an entry point for a photochemical event that deconstructs the azine into a nitrile-containing diene primed for Diels–Alder cycloaddition. The method displays a wide functional group tolerance allowing for the streamlined preparation of complex arene building blocks. We have demonstrated how this approach is amenable to the late-stage ring replacement of a variety of 4-substituted pyridine drug derivatives and allows for the rapid assembly of densely functionalized molecular libraries. We hope that the commercial availability of pyridines and the broad scope demonstrated here will make this strategy of use to industrial and academic end-users.
Methods
General procedure for the synthesis of aminopentadienenitriles
Pyridine-N-oxide (0.1 mmol, 1 equiv.) was added to a microwave vial, which was then capped with a Supelco aluminium crimp seal with a septum (PTFE/butyl), evacuated and purged with argon (×3). Degassed CH3CN (5 ml, 0.02 M) was added followed by morpholine (88 μl, 1 mmol, 10 equiv.) and the solution was degassed with a stream of argon for 20 s. The microwave vial was then irradiated with 310 nm light in a Helios photoreactor for 15 h. The cap was then removed, and the solvent was evaporated to give a crude residue which was purified via column chromatography eluting on neutral Al2O3 to give the pure aminopentadienenitrile.
General procedure for the Diels–Alder cycloaddition
1 (27 mg, 0.1 mmol, 1 equiv.) was added to a microwave vial followed by dienophile (0.4 mmol, 4 equiv.), if solid. The vial was capped with a Supelco aluminium crimp seal with a septum (PTFE/butyl), evacuated and purged with argon (×3). Toluene (0.1 ml, 1 M) and dienophile (0.4 mmol, 4 equiv.), if liquid, were added. The sealed vial was then heated at 120 °C for 15 h. The cap was then removed, and the solvent was evaporated to give a crude residue which was purified via column chromatography eluting on silica gel to give the pure benzonitrile.
General procedure for one-pot arene synthesis
Step 1 (ring opening)
Pyridine-N-oxide (0.1 mmol, 1 equiv.) was added to a microwave vial, which was then capped with a Supelco aluminium crimp seal with a septum (PTFE/butyl), evacuated and purged with argon (×3). Degassed CH3CN (5 ml, 0.02 M) was added followed by morpholine (88 μl, 1 mmol, 10 equiv.) and the solution was degassed with a stream of argon for 20 s. The microwave vial was then irradiated with 310 nm light in a Helios photoreactor for 15 h. The cap was then removed, and the solvent was evaporated to give the crude aminopentadienenitrile which was dried under high vacuum to remove trace volatiles to give a crude residue which was used directly in the next step without further purification.
Step 2 (cycloaddition)
To the crude aminopentadienenitrile from step 1, toluene (0.1 ml, 1 M) and ethyl propiolate (41 μl, 0.4 mmol, 4 equiv.) were added. The vial was capped with a microwave vial lid and the reaction was then heated in a microwave (μW) reactor at 150 °C for 2 h. The cap was then removed, and the solvent was evaporated to give a crude residue which was purified via column chromatography eluting on silica gel to give the pure benzonitrile.
Responses