Antibody-functionalized MXene-based electrochemical biosensor for point-of-care detection of vitamin D deficiency
Introduction
Despite the abundant sunshine in Saudi Arabia and across the Middle East and North Africa (MENA) region, around 80% of the population, spanning various age groups, suffers from vitamin D (Vit-D) deficiency. The deficiency rate in North America is approximately 24–41%, while in Europe, it ranges from 30–60%1,2,3,4. The variation in Vit-D deficiency can be attributed to cultural and lifestyle practices, genetic predispositions, intense heat, and varying skin tones5. Based on a clinical study of Vit-D deficiency in the central region of Saudi Arabia, Al Daghri et al. reported that approximately 65% of the population suffered from deficiency, while 16% suffered from severe deficiency1. Vit-D is a fat-soluble seco-steroid hormone and an essential vitamin for the body to regulate calcium and phosphorus6. These nutrients are necessary to keep bones, teeth, and muscles healthy. In principle, Vit-D can be classified as either 25(OH)D2/ergo-calciferol (Vit-D2) or 25(OH)D3 /cholecalciferol (Vit-D3). The latter metabolic form, Vit-D3, is obtained from non-fortified food, fortified food, and supplements, while Vit-D2 can be obtained from non-fortified food such as mushrooms and supplements. Notably, Vit-D deficiency (defined by levels below 20 ng mL−1) and severe deficiency (defined by levels below 10 ng mL−1) are considered severe health issues that can lead to complications and diseases such as schizophrenia, coronary heart diseases, hypertension, Parkinson’s disease, Alzheimer’s disease, and skeletal deformation7.
Consequently, timely and facile detection of Vit-D levels can significantly and positively impact the healthcare and quality of life for people in the MENA region and beyond. While many strategies have been proposed in the literature for detecting Vit-D levels based on enzyme-linked immunosorbent assays (ELISA), surface-enhanced Raman spectroscopy8, mass spectrometry9, surface plasmon resonance10, chromatography11,12, and radioimmunoassays12, most of these strategies are expensive, time-consuming, and require sophisticated instrumentation with expert operators. For example, Woochang K. et al. developed an Ag Danville-modified SERS aptasensor for Vit-D detection, but this approach required expensive instrumentation with expert personnel, making it unsuitable for use in decentralized locations13. Electrochemical sensors present notable advantages for this application, as they can be designed to offer portability, sensitivity, and ease of operation14. Shikha et al. developed an impedance-based electrochemical biosensor based on graphene quantum dot-Au hybrid nanoparticles for aptamer-based Vit-D3 detection15. Kun Men et al. demonstrated the electrochemical detection of Vit-D2 and Vit-D3 by modifying a glassy carbon electrode (GCE) with an Au Pd bimetal. However, the latter electrode configuration was unsuitable for point-of-care use, and the incorporation of noble metals made this strategy costly16,17,18,19. Therefore, given the importance of Vit-D monitoring and the lack of available fit-for-purpose technologies, there is a pressing need for the development of point-of-care biosensing devices that are rapid, reliable, user-friendly, affordable, sensitive, specific, equipment-free, and deliverable at decentralized locations10,20,21,22.
Recently, two-dimensional (2D) nanomaterials such as graphene and MoS2 have demonstrated significant promise as transducers in advanced electrochemical sensors and biosensors23,24. Among them, MXenes have emerged as a rapidly growing family of 2D transition metal carbide and carbonitride materials that possess a myriad of physical and chemical advantages enabling them to be exploited in a range of applications25, including energy storage, electromagnetic shielding, physical sensing, biosensing26,27, and electronics28,29. Notably, MXene’s excellent metallic conductivity of up to 24000 S/cm30, abundance of tunable surface functional groups, 2D-like planar structure, and biocompatibility make them especially promising in biosensing applications23,25. We have recently developed a MXene-based biosensor by directly clicking peptide nucleic acid probes on the MXene surface for the detection of nucleic acids, specifically microRNA13. Previous work has also demonstrated the development of MXene immunosensors based on silanization of antibodies on MXene surfaces for the electrochemical detection of proteins including the cancer biomarker carcinoembryonic antigen (CEA)31, and the allergenic buckwheat protein BWp1632. However, MXene has rarely been explored in vitamin sensing, with only one reported case by Rijo et al. that demonstrated the electrodeposition of manganese dioxide-inorganic phosphate (MnO₂-PI) on MXene nanosheets for the catalytic-detection of vitamin B6. This approach did not use any bioreceptor, thereby lacking the demonstration of selectivity or any validation with biological samples33. This study leverages the superior electrochemical and physicochemical properties of functionalized MXene in developing and validating the application of MXene in vitamin D biosensing, with thorough characterization and validation studies highlighting its applicability and potential for clinical translation.
Herein, we present the design, development, and validation of a robust and point-of-care electrochemical biosensor for vitamin D3 (hereafter referred to as Vit-D) based on biofunctionalized MXene, offering clinical sensitivity and high specificity without requiring amplification, enzymes or complex nanocomposite materials. To serve as a portable and robust electrode material, we have implemented laser-induced graphene (LIG) due to its single-step fabrication process, highly porous structure, excellent conductivity, easy patterning, and cost-effectiveness compared to glassy carbon electrodes (GCE) or other screen-printed carbon electrodes (SPCE)34. Following the single-step LIG fabrication, the preparation of our proposed platform involves (1) electrostatic interactions between the negatively-charged terminal groups of MXene and positively-charged functional groups of polyethyleneimine (PEI) and (2) covalent bonding to the anti-Vit-D antibody using glutaraldehyde (GA) as a crosslinking agent between the amino groups of PEI and amino groups of the antibody (Scheme 1). Following thorough materials characterization studies to confirm successful functionalization, we characterized the analytical performance of the Vit-D biosensor. The platform exhibited a wide detection range (0.1 to 500 ng mL−1), encompassing levels indicative of clinical deficiency, insufficiency, sufficiency, and toxicity. Notably, the limit of detection reached an impressive 1 pg mL−1 without the need for enzymes, non-isothermal conditions, or complex amplification strategies owing to the outstanding material and electrochemical properties of the functionalized MXene. Given its impressive analytical performance and suitability for point-of-care testing, we believe this platform offers significant promise in developing next-generation biosensors implemented in critical healthcare, environmental monitoring, and food safety applications.
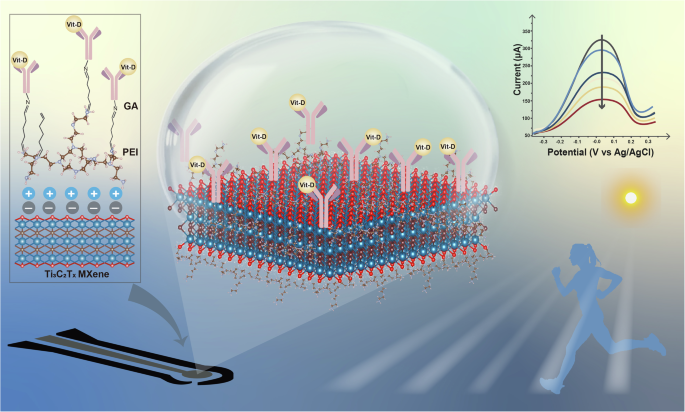
Schematic illustration of the antibody-functionalized MXene-based electrochemical sensor for Vit-D detection.
Results and discussion
MXene synthesis, functionalization with PEI, and characterization
MXene nanosheets were prepared following a well-established protocol from our previous publications (additional details are provided in the Materials and Method Section in the Supplementary Information)35. In a typical experiment, Ti3AlC2 MAX phase powder was etched using an etchant solution to produce accordion-like Ti3C2Tx (MXene) nanosheets. These nanosheets were then mixed with an intercalator to facilitate intercalation and delamination of the MXene nanosheets. To enable covalent functionalization with the antibody bioreceptor, we deposited a polyethyleneimine (PEI) layer through electrostatic interactions to introduce amine functional groups. Briefly, MXene nanosheets were redispersed in Poly(ethyleneimine) solution at the ratio of (3 mL: 100 µL), respectively, and stirred for 2 h at room temperature to get Amino-MXene (Supplementary Information, Materials and Method Section). The schematic representation for the synthesis of MXene, Amino-MXene, and single layer-MXene are shown in Fig. 1a, b, and Supplementary Fig. S1, respectively.

Schematic representation of the synthesis route for a MXene, and b Amino-MXene (Inset: photograph of MXene, and Amino-MXene). c TEM (inset: selected area electron diffraction (SAED) pattern) of MXene nanosheet; d AFM characterization of a MXene monolayer. e Zeta potential measurement of MXene and Amino-MXene, prepared in DI water. f XRD spectra of MAX phase, MXene and Amino-MXene. g Raman spectra of MXene and Amino-MXene.
The quality of the pristine MXene sheets was assessed using X-ray diffraction (XRD) analysis, transmission electron microscopy (TEM), and atomic force microscopy (AFM) (Bruker, Dimension Icon SPM). The effective delamination of Ti3C2Tx MXene flakes was confirmed through TEM (Fig. 1c) and AFM (Fig. 1d) analyses. TEM micrographs of the MXene flakes are shown in Fig. 1c exhibiting a well-defined 2D nanosheet morphology31. The selected area electron diffraction (SAED) pattern of the as-prepared MXene sample, depicted in the inset of Fig. 1c, revealed a hexagonal symmetry characteristic of its structure. This observation suggested that the MXene powder maintained the single-crystal nature and symmetry of its parent phase, Ti3AlC236,37.
Additionally, AFM measurements were conducted to analyze the surface morphology and estimate the thickness of a single layer of the synthesized MXene nanosheets. Figure 1d depicts the typical morphology of a mono- and few-layered of MXene nanosheet on a glass substrate, with a 1.7 nm thickness, affirming the attained proper delamination38. Moreover, the flake size distribution of MXene was characterized by dynamic light scattering (DLS), exhibiting an average flake size of (3.1 ± 0.5) µm as shown in Supplementary Fig. S2.
Next, we analyzed the surface charge and colloidal stability of both the pristine MXene and functionalized Amino-MXene suspensions using Zeta potential (ZP) measurements, as shown in Fig. 1e. Due to the negatively charged surface functional groups (-OH, -O, -F), bare MXene exhibited a ZP of −39.6 mV in water, indicating excellent colloidal stability with high affinity for positive ions/charges39. However, after functionalization with PEI, the ZP increased to 40.9 mV, indicating successful electrostatic attraction between cationic PEI and anionic MXene nanosheets.
Following the processes of etching, intercalation, and subsequent delamination, the distinct (002) peak was downshifted from its typical 2θ angle of ~9.4° for the parent MAX phase to a lower 2θ angle of approximately ~7.3° indicating increasing in d-spacing by the surface terminating groups (Tx, i.e.,= —OH, —F, —O, etc.) during the etching process as shown in Fig. 1f38.
Moreover, the featured gradual broadening of the (002) peak throughout the processing stages, indicates a less ordered layer-by-layer stacking, i.e., another indication of the proper delamination and the reduction thickness of the MXene layers40,41. However, after surface modification with PEI polymer, the intensity of the sharp 002 peaks of MXene decreased, accompanied by a broadening of the peak observed between 18-28° degrees, indicating the presence of amorphous PEI polymer as shown in Fig. 1f42,43.
Raman spectroscopy using laser radiation with a wavelength of 532 nm was next exploited as a characterization method for examining the surface chemistry, interlayer spacing, and number of defects of pristine and PEI-functionalized MXene44. Fig. 1g illustrates the Raman spectra showcasing characteristic peaks of MXene at 202, 277, 373, 622, and 729 cm−1, which correspond to the following vibration modes: A1g of Ti3C2O2, Eg of Ti3C2(OH)2, Eg of Ti3C2O2, Eg of Ti3C2(OH)2, and A1g of Ti3C2O2, respectively indicating successful synthesis of MXene44,45. However, after functionalization with PEI, highly intense Raman bands were observed in the region of 1300–1600 cm−1, which corresponds to the -NH bending vibration of PEI46,47.
MXene functionalization with anti-vit-D antibody and associated characterization.
Following the formation of the Amino-MXene sample, the antibody functionalization process involved the introduction of a bivalent crosslinker called glutaraldehyde (GA) to enable two covalent bonds, firstly, between PEI and GA’s aldehyde (—CHO) groups to form LIG-Amino-MXene-GA, and secondly, between GA and the amine groups of the antibody to form LIG-Amino-MXene-GA-Ab (Additional details on the sensor design, and fabrication, and preparation can be found in the Supplementary Information, as shown in Supplementary Fig. S3). Successful functionalization was validated through X-ray photoelectron spectroscopy (XPS) and energy dispersive X-ray (EDX) analysis.
To characterize the changes in the surface chemistry of the MXene sheets during each step of the functionalization process, high-resolution XPS analysis was conducted, and the corresponding spectra of the C1s and N1s core levels were acquired. Figure 2a shows that most of the carbon content ( ~95%) in the LIG+Amino-MXene sample, which is centered at a binding energy of 286.6 eV, can be attributed to carbon-nitrogen bonds such as C-NH/NH2 and C-N-C of the PEI layer. The remaining 5% of the carbon distribution located at 284.8 eV and 288.4 eV can be attributed to the PEI C-C and C-O bonds, respectively. Figure 2b shows the N 1 s spectrum of the LIG+Amino-MXene sample, where 91% nitrogen distribution was detected at 400.3 eV.
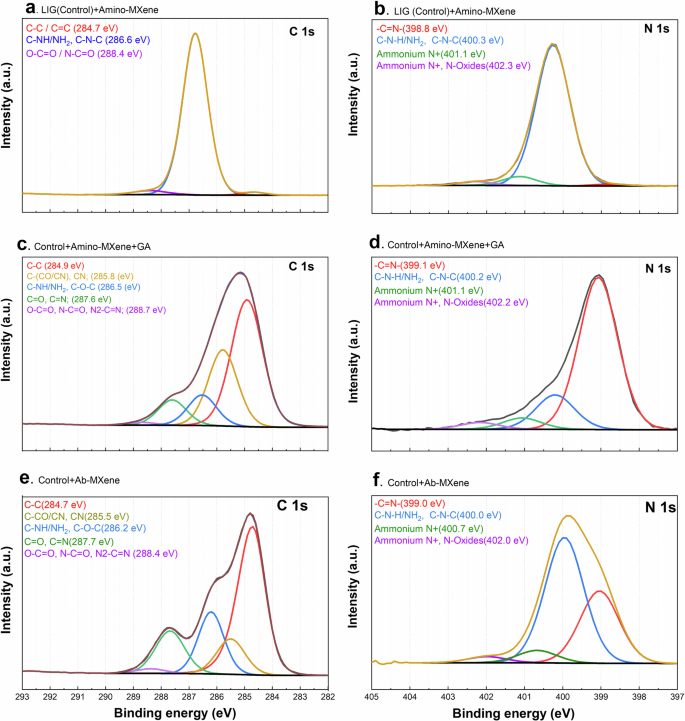
High-resolution XPS spectra for (C 1 s and N 1 s) core levels of a, b LIG (control)+Amino-MXene, c, d control+Amino-MXene+GA, and e, f LIG+Ab-MXene samples.
This nitrogen peak was attributed to C-NH/NH2 and C-N-C48, while the higher binding energy peaks above 401 eV were assigned mainly to protonated nitrogen species. The small shoulder peak at 398.8 eV, contributing to 1% of the nitrogen content, was assigned to the imine group (—C = N—)49. The LIG+Amino-MXene sample was composed of 65% carbon, 31% nitrogen, and 4% oxygen based on both the C 1 s and N 1 s spectra, which accords well with the theoretical expectations of C and N in the PEI structure. Changes in the C 1 s spectra could also be used to monitor changes in the material following GA immobilization, as shown in Fig. 2c. Obviously, the C-C bonds from the GA structure were detected at 285.8 eV with C-N bonds and carbon atoms adjacent to the C = O groups located around50. The peaks at 286.6 eV, representing C-NH/NH₂ groups were reduced significantly—from 95% in the LIG-Amino-MXene sample to just 10% in the (LIG + Amino-MXene + GA) sample. Meanwhile, a new peak associated with C = O and C = N groups appeared at 287.6 eV51,52, validating the successful —C = N chemical bonding formation between PEI and GA. Furthermore, the imine group (—C = N—) peak located at 398.8 eV was significantly enhanced after immobilizing GA on the PEI layer, contributing to 74% of the nitrogen distribution, as shown in Fig. 2d. However, the peak attributed to C-NH/NH2, C-N-C located at 400.2 eV, had a pronounced drop, demonstrating the formation of numerous —C = N— bonds between PEI and GA compounds. The components detected at higher binding energies and assigned mainly to protonated nitrogen species remained unchanged.
After conjugating the antibody to the GA crosslinker, XPS analysis of the C 1 s spectrum (Fig. 2e) showed that the component at 285.5 eV, assigned to CN and carbons adjacent to CO and CN groups, decreased compared to the previous sample. In addition, the species attributed to C-NH/NH2 (at 286.2 eV) and C = O / C = N groups (at 287.7 eV) increased along with the component detected at 288.4 eV, which was ascribed to carbons bound to more than one O and N atom or bound at the same time to O and N (carboxylic and amide groups among other possibilities). These data evidence the clear signature of antibodies as their chemical structure contains C-NH, C-NH2, carboxylic (COOH), and amide (N-C = O) groups, all of which were detected with higher intensities compared to the previous non-functionalized sample. Moreover, N 1 s analysis indicated a decrease in the 399 eV peak assigned to —C = N bonds, as shown in Fig. 2f, while the main component detected at 400 eV, ascribed mainly to C-NH/NH2, became more predominant, accounting for 58 at%. However, the N 1 s peak attributed to —C = N was still detected with significant intensity (nearly 33 at%), suggesting the formation of —C = N— bonds between GA and antibodies on the sample surface.
In addition to XPS analysis, the compositional and morphological changes of the sample materials were monitored at various stages of modification using energy-dispersive X-ray (EDX) analysis and scanning electron microscopy (SEM) (Fig. 3 and Supplementary Fig. S4, respectively). Initially, bare LIG displayed a uniform distribution of carbon (C), oxygen (O), and nitrogen (N) (Fig. 3a). Upon the introduction of MXene to the LIG, titanium (Ti) and fluorine (F) signals appeared (Fig. 3b). The subsequent introduction of functionalized Amino-MXene intensified the nitrogen (N) mapping compared to bare LIG and MXene samples, which was attributed to the abundance of nitrogen in the amino (—NH2) groups of PEI (Fig. 3c). Crosslinking with GA has led to a further increase in C and O signals and an evident decrease in the Ti signal, as GA molecules containing aldehyde groups covered the LIG-Amino-MXene surfaces (Fig. 3d). Finally, upon antibody immobilization onto the LIG-Amino-MXene-GA surface, a slight increase in oxygen mapping intensity was observed due to the presence of oxygen components in the antibody structures (Fig. 3e).

a LIG, b LIG-MXene, c LIG+Amino-MXene, d LIG+Amino-MXene+GA, e LIG+Ab-MXene.
Electrochemical Characterization and DFT Simulation of the Amino-MXene Structure
The stepwise modification of the sensor (Fig. 4a) was monitored and electrochemically characterized using cyclic voltammetry (CV) in the presence of PBS solution containing 5 mM [Fe(CN)6]3-/4- in the potential window of −0.3 to 0.4 V at a 50 mV/s scan rate (Supplementary Fig. S5a). Firstly, we characterized the electrochemical behavior of bare LIG (black curve, Supplementary Fig. S5a), which demonstrated the expected redox peaks for the chosen redox reporter. After adding pristine MXene to the LIG surface (red curve, Supplementary Fig. S5a), we observed a slight (~13%) decrease in the oxidation peak current, attributed to successful coating of LIG’s porous structure with the MXene flakes, which may slightly reduce the sensor’s electrochemically active surface area.

a Schematic illustration of the stepwise preparation of the Vit-D sensor. Characterization of the stepwise modification of the sensor using b CV in the potential window of −0.3 to 0.4 V at 50 mV/s scan rate (5 scans were acquired per condition, with the last scan plotted), and c Nyquist plot at different stages of the sensor modification (inset: shows the zoomed -in view of the x-axis intercept, and Randle’s equivalent circuit showing Rs (solution resistance), Rct (charge transfer resistance), Cdl (interfacial capacitance), and W (Warburg resistance); 3 scans were acquired per condition (with the last scan plotted), d Projected Density of States (PDOS) of the PEI adsorbed on the MXene surface, e optimized geometry of PEI molecules on the MXene (100) surface, and f charge density difference plot when PEI is adsorbed on MXene. g Diffusion-controlled analysis using CV in the presence of 1X PBS solution containing 5 mM [Fe(CN)6]3-/4-, where all experiments were repeated 3 times. h The corresponding calibration curve obtained from the peak currents versus square root of the scan rate.
After drop-casting the aminated MXene, Amino-MXene, onto the LIG electrode, the sensor’s oxidation peak current was enhanced by approximately 52% (blue curve, Fig. 4b) compared to the bare LIG-MXene electrode. This enhancement was attributed to the cationic nature of PEI, which facilitates electron transfer between the redox species and the electrode through electrostatic attraction53. Moreover, the additional polymeric layer of PEI on the electrode surface may increase the effective surface area available for redox reactions. Next, we acquired cyclic voltammograms of the sensor after the covalent functionalization of the antibody through the GA crosslinker. We observed a 16% (green curve, Fig. 4b) decrease in oxidation peak current, which can be attributed to the large proteins (antibodies) physically blocking conductive pathways, creating an insulating effect and steric hindrance on the sensor surface54. As expected, the redox peak currents further decreased by 33% (purple curve, Fig. 4b), after treating the surface with a blocking agent, namely BSA, which blocks the sensor surfaces which are not functionalized with an antibody53,54.
In addition to cyclic voltammetry, we next sought to electrochemically analyze each modification step using electrochemical impedance spectroscopy (EIS), as shown in Fig. 4c (Further details on the EIS measurement can be found in the Supplementary Information). The bare LIG electrode exhibited a charge transfer resistance (Rct) of 405 Ω (black curve, Supplementary Fig. S5b), which increased to 463 Ω (red curve, Supplementary Fig. S5b) after modification with MXene, likely due to the coverage of the porous LIG network by MXene flakes, consequently reducing the sensor’s active surface area43. However, replacing MXene with Amino-MXene resulted in a significantly lower Rct value of 136 Ω (blue curve, Supplementary Fig. S5b), indicating faster electron transfer between the redox species and the electrode through electrostatic interactions. After antibody immobilization, the Rct increased again to 152 Ω (green curve, Fig. 4c), suggesting the physical blocking of conductive pathways via large proteins like antibodies and the creation of steric hindrance (Yunhua Lu et. al. 2020)44. Finally, after BSA blocking, the Rct value further increased to 302 Ω (purple curve, Fig. 4c), as expected53,54. The peak currents and charge transfer resistance at various stages of surface modification of the vitamin D sensor, as extracted from Fig. 4b and Fig. 4c, are summarized in Supplementary Table S1.
Before proceeding with analytical experiments, we first sought to investigate and understand the favorable and high electrochemical performance of the Amino-MXene sample. For that, pristine MXene and PEI interactions were simulated through Density Functional Theory (DFT) using the Vienna ab initio simulation package (VASP)55,56. For our purposes, only one unit of PEI was considered on the MXene surface to study the geometry of PEI adsorption during the simulation, which was performed at the generalized gradient approximation level using the Perdew-Burke-Ernzerhof (PBE) functional, with Van der Waals dispersion effects considered at the DFT-D3 level57,58. Optimization employed the Broyden–Fletcher–Goldfarb–Shanno (BFGS) method until the maximum force reached was below 0.02 eV/Å. A plane-wave cut-off of 400 eV was set, employing a repeated-slab method to ensure a minimum of 15 Å vacuum between the slab and organic molecule. Spin-unpolarized slab calculations were conducted for a monolayer of MXene using an 8 × 8 supercell (dimension nearly 24 Å along x and y directions) and a 2x2x1 Monkhorst-Pack k-point mesh. Density of states (DOS) calculations were performed with a denser 3x3x1 k-point mesh.
The interface between MXene and PEI was analyzed by the difference in charge density between the hybrid and individual components as
where ({rho }_{{{rm{MXene}}}/{{rm{PEI}}}}),(,{rho }_{{PEI}}), and ({rho }_{{MXene}}) represent the total (PEI and MXene) and individual component charge densities, respectively, while ({rho }_{{diff}}) is the charge density difference at the interface. Figure 4d shows the projected density of states (PDOS) of the Ti3C2Tx-PEI interface. The optimized geometry of the PEI molecule on the MXene (100) facet is displayed in Fig. 4e, where the binding energy between the MXene and the PEI was found to be −4.13 eV, as shown in the charge density difference plot when PEI is adsorbed on the Ti3C2Tx MXene. Such a high binding energy between Ti3C2Tx and PEI corresponds to a charge density rearrangement at the interface between PEI and MXene, as revealed by charge difference analysis (Fig. 4f). This important observation can shed light on the observed favorable electrochemical performance of the Amino-MXene sample, which can consequently lead to high sensitivity and improved signal-to-noise ratio in the electrochemical sensor59.
To probe the diffusion state of the sensor, CVs were acquired at different scan rates (from 5 to 80 mV/s) as shown in Fig. 4g. The peak currents were plotted against the square root to the scan rate, which showed a linear relationship for both anodic and cathodic peak currents (Fig. 4h), indicating diffusion-controlled reactions at the solid-liquid interface of the sensor and electrolytes60.
Sensor’s analytical performance: electrochemical detection of Vit-D
Following the electrochemical characterization of our Vit-D biosensor, we next sought to characterize its analytical performance using DPV analysis. Figure 5a shows a schematic illustration of the electrochemical detection of Vit-D using our biosensor. Under the optimized electrochemical parameters, we demonstrated Vit-D detection on the disposable, low-cost antibody-functionalized MXene sensor using DPV (from −0.3 V to 0.3 V) at the various concentrations of Vit-D in 1X PBS solution containing 5 mM [Fe(CN)6]3-/4- as shown in Fig. 5 b, c. The peak current in the DPV measurements decreased proportionally with increasing concentrations of Vit-D (ranging from 0.1 to 500 ng mL−1). This trend was attributed to the hindrance of electrochemical reactions caused by the formation of immunocomplexes on the sensing surface61. To assess the sensitivity of the biosensor, we generated calibration curves by plotting control-subtracted peak currents against Vit-D concentration, as depicted in Fig. 5c. The calibration curve showed a linear response (in log-logscale) with remarkable sensitivity and an excellent limit of detection (LOD) of 1 pg mL−1 with a coefficient of determination (R²) of ≥ 0.99. The LOD was calculated as three times the standard deviation of the control condition. The curve covered a wide dynamic range from (0.1–500) ng mL−1, which includes the clinical ranges for Vit-D deficiency, insufficiency, sufficiency, and toxicity. Notably, our sensor’s sensitivity surpassed that of recently reported similar electrochemical biosensors, as detailed in the Supplementary Information (Table S2). Based on the proposed Vit-D detection sensor strategy, our disposable, low-cost antibody-functionalized MXene sensor was able to achieve a wide dynamic range (spanning five orders of magnitude) and an ultralow LOD of 1 pg mL−1 relevant to clinical applications.

a Schematic illustration representing the Vit-D biosensor’s electrochemical detection strategy. b Differential pulse voltammetry (DPV) scans of the Vit-D sensor at different concentrations (0, 0.1, 1, 10, 100, and 500 ng mL−1) of Vit-D in 5 mM [Fe(CN)6]3-/4- in 1X PBS solution. c Calibration curve (log of normalized current versus log of Vit-D concentration) of the Vit-D biosensor. Error bars indicate the standard error of the mean for N ≥ 6 data points collected from two or three individual biosensors with three DPV scans acquired per concentration. d Interference or specificity test of the Vit-D sensor in the presence of non-target analytes: glucose (7 mM), Vit-C (1 mM), Vit-B12 (300 pg mL−1 or 2.2 zM), miR-141 (10 fM) and Vit-D (25(OH)D3), (10 ng mL−1 or 0.0265 µM) in 5 mM [Fe(CN)6]3-/4- in 1X PBS solution (**** indicates p < 0.0001). eData was collected from one sensor per analyte type (with three DPV scans per sensor) and signalswere compared to that from the target Bit-D, normalized to 100%. e Reproducibility test of the Vit-D sensors which were all incubated with the same concentration of target Vit-D (10 ng mL−1) tested with 5 mM [Fe(CN)6]3-/4- in 1X PBS solution. Signals from Sensors 2-5 were compared to the signal from Sensor 1, normalized to 100%.
Sensor’s performance under physiological conditions and reproducibility
In addition to device sensitivity, the selectivity of a biosensor is a crucial parameter to investigate due to the presence of other similar analytes circulating in biofluids, including nucleic acids, metabolites, glucose, and even other vitamins.
Under clinical testing conditions, the biosensor can be exposed to other non-target biomolecules, which may compete with or hinder our sensor performance. Thus, to investigate our biosensor under these conditions, we prepared solutions of four non-target biomolecules at their expected physiological conditions, namely glucose (7 mM), Vit-C (1 mM), Vit-B12 (300 pg mL−1 or 2.2 zM), and miR-141 (10 fM), and measured their individual DPV response as shown in Fig. 5d. Notably, as a comparison, we prepared our target biomolecule, vitamin D, under “deficiency” conditions (specifically, 10 ng mL−1 or 26 nM) to investigate whether the biosensor can detect minimal levels of vitamin D compared to physiological concentrations of other non-target biomolecules. The results were normalized (against the target vit-D response, which was set to 100%), and showed only a minor (<20%) signal arising from the non-target biomolecules, indicating high selectivity for Vit-D (unpaired, one-tailed t-test and Welch corrections, with p < 0.0001 considered statistically significant).
Another critical property in point-of-care devices is their reproducibility. To investigate this, five identical LIG-Amino-MXene-GA-Ab sensors were prepared for a Vit-D detection test. Following the same optimized protocol using DPV analysis, the response of all sensors to 10 ng mL−1 of Vit-D was compared and showed good reproducibility, as shown in Fig. 5e. Sensor 1 was selected as the control, whose response was normalized to 100%. Compared to this, Sensors 3, 4, and 5 exhibited slight changes with deviations of 8.4%, 10%, and 3.3%, respectively. However, Sensor 2 showed a substantial 15% deviation compared to Sensor 1, which may be attributed to human errors in handling and sample preparation. These findings highlight the good reproducibility of our Vit-D sensor, which is a crucial characteristic for ensuring successful translational development. Finally, we investigated the performance of our biosensor using complex biological samples, specifically human serum, spiked with known concentrations of our target Vit-D. Notably, our platform was able to successfully detect the target from serum samples; however, we observed significant reductions in the analytical signal, attributed to surface biofouling effects (Supplementary Fig. S6). Consequently, our future work will focus on developing robust antifouling strategies for our biosensing platform.
Conclusions
In this work, we demonstrate the development and validation of a facile, robust, and enzyme-free electrochemical biosensor for Vit-D detection based on advanced biofunctionalized 2D MXene. A highly efficient and ultrasensitive sensing platform was achieved by functionalizing Ti3C2Tx MXene with anti-Vit-D antibodies, facilitated by amination with PEI and subsequent covalent bonding. The functionalization process was rigorously validated through XPS and EDX characterization studies, while simulations via DFT contributed to a deeper understanding of the device’s superior electrochemical performance. Electrochemical characterization studies of the sensor’s analytical performance revealed its remarkable and clinically relevant sensitivity, with a limit of detection as low as 1 pg mL−1, coupled with a broad dynamic range spanning from 0.1 ng mL−1 to 500 ng mL−1. Importantly, this dynamic range covers the clinically relevant spectrum of Vit-D concentrations from clinical deficiency to toxicity, offering great promise for rapid and accurate detection of Vit-D levels in clinical settings. Overall, this work underscores the potential of MXene-based electrochemical biosensors in enabling highly sensitive and specific detection of biomarkers of disease, paving the way for advancements in medical diagnostics and personalized healthcare at the point of need.
Responses