Brain O-GlcNAcylation: Bridging physiological functions, disease mechanisms, and therapeutic applications
Introduction
Most eukaryotic proteins undergo post-translational modifications (PTMs), such as phosphorylation, methylation, ubiquitylation, acetylation and glycosylation [1, 2]. Among these, O-linked β-N-acetylglucosaminylation, commonly referred to as O-GlcNAcylation, is a dynamic glycosylation that occurs on serine or threonine residues of proteins [3, 4]. In 1983, while analyzing sugar chains in blood lymphocyte membranes, Hart et al. unexpectedly discovered a monoglycosylation modification distinct from traditional N- and O-glycan modifications [3]. This modification involved a monosaccharide, acetylated at the amino terminus, with glucosamine attached to the hydroxyl group of serine or threonine, which led to its designation as O-GlcNAc modification [3]. In the subsequent decade, the Hart laboratory identified β-N-acetylglucosamine glycosyltransferase (O-GlcNAc transferase, OGT), the enzyme responsible for O-GlcNAcylation, and aminoglycoside hydrolase (O-GlcNAcase, OGA), which catalyzes the deglycosylation of β-N-acetylglucosamine [5, 6]. Since the 1990s, many studies have discovered that many O-GlcNAcylation sites directly compete with phosphorylation sites [7]. Recently, the rapid development of proteomics and the widespread application of mass spectrometry have facilitated the identification of an increasing number of O-GlcNAc-modified proteins and their specific modification sites. In recent years, advances in omics technologies and modern detection methods have provided deeper insights into the role of O-GlcNAc modification in various biological processes. Owing to its dynamic nature and spatiotemporal specificity, the concept of O-GlcNAc glycomics (O-GlcNAcomics) have been introduced, marking a significant advancement in the O-GlcNAc modifications [8].
Since its initial identification in the 1980s, O-GlcNAcylation has evolved as a pivotal player in numerous cellular functions. This modification is orchestrated by two opposing enzymes: OGT and OGA. Mechanistically, OGT attaches a GlcNAc moiety to the target protein [9, 10], while the hydrolase OGA catalyzes the removal of this sugar moiety [11], allowing the levels of O-GlcNAcylation to dynamically adjust in response to cellular processes [12]. Extensive research has revealed that O-GlcNAcylation is a ubiquitous PTM involved in a wide array of cellular activities. These include gene regulation and expression [13,14,15], metabolic activities [16], cell cycle regulation [17, 18], nutrient sensing [19], and responses to stress conditions [20]. Notably, O-GlcNAcylation is observed in a diverse array of cells, from viruses and bacteria to T and B cells, as well as neural cells in the brain [21, 22]. This modification is prevalent across various subcellular compartments, extending beyond the cytosol and nucleus to include cellular organelles [23, 24]. Additionally, membrane proteins have been recognized as targets of O-GlcNAcylation at their cytosolic tail [23, 25]. Collectively, these findings underscore the widespread influence of O-GlcNAcylation in regulating key aspects of cellular physiology.
O-GlcNAcylation is a prevalent PTM in the central nervous system (CNS), which is critical for the development, physiological function, and the pathogenesis of brain diseases by modulating diverse downstream proteins [26, 27]. Notably, various proteins commonly present at synapses are O-GlcNAcylated [28]. It has been demonstrated that numerous proteins critical for the structure and function of neurons, astrocytes, and synapses undergo O-GlcNAcylation [29]. Meanwhile, O-GlcNAcylation is linked to neurogenesis [30], axonal and dendritic growth and function [31, 32], as well as the regulation of neuronal metabolism [33], cognitive function [34, 35], inflammatory activation and stress response of astrocytes [35, 36]. Proper physiological functioning requires a balanced state of O-GlcNAcylation, and any deregulation can predispose to pathological conditions. Dysregulation of O-GlcNAcylation has been associated with a spectrum of CNS disorders, encompassing neurodevelopmental disorders [37,38,39,40], neurodegenerative diseases [41,42,43,44,45], neuropsychiatric disorders [36, 46, 47], and CNS injuries [48,49,50]. Thus, understanding the mechanisms of O-GlcNAcylation and its impact on neuronal health remains a critical area of neuroscientific research.
In this review, we endeavor to provide an extensive review of the critical roles played by O-GlcNAcylation in the developmental and physiological functioning of the CNS. Additionally, we examine the essential role of O-GlcNAcylation in pathological conditions, with a particular focus on its neuroprotective effects under such conditions. Finally, we explore the feasibility of targeting O-GlcNAcylation as a novel therapeutic approach for treating these neurological conditions.
Neurodevelopmental functions of O-GlcNAcylation in the brain
In the complex milieu of neural development, O-GlcNAcylation assumes a critical regulatory role. This modification orchestrates a plethora of cellular processes, particularly within the cerebral domain. Recent scholarly investigations have delineated the multifarious roles of O-GlcNAc, thereby underscoring its paramount significance in the ontogeny of the nervous system.
The role of O-GlcNAc in neurogenesis
Neural stem/progenitor cells (NSCs/NPCs) are the primary determinants of neurogenesis, and O-GlcNAc plays a crucial role in the proliferation and maintenance of these cells. Targeted deletion of Ogt in NSCs during early embryonic development (E13.5) leads to impaired cortical development and significant degenerative changes in the murine brain postnatally. This deletion results in diminished proliferation of embryonic NSCs and impaired migration of newly generated neurons, accompanied by a notable elevation in endoplasmic reticulum (ER) stress levels within mutant neurons [30]. In a human neuron model, O-GlcNAc governs the development and maturation of cortical neurons originating from human induced pluripotent stem cells by modulating primary cilium length. Notably, O-GlcNAc exerts a negative regulatory influence on cilium length in differentiated cortical neurons [51]. Additionally, during human embryonic stem cell differentiation, O-GlcNAcylation of RING1B contributes to the maintenance of pluripotency via the regulation of the polycomb repressive complex 1 (PRC1), a pivotal epigenetic repressor [52]. Furthermore, inhibition of OGT with chemicals during neural induction of human embryonic stem cells, achieved through treatment with peracetylated 5-thio-N-acetylglucosamine (Ac4-5SGlcNAc), accelerates differentiation along the neuronal lineage, thereby offering insights into the dynamic molecular mechanisms underpinning neuronal development [53]. Within adult neural stem or progenitor cells, Ogt plays a pivotal role in governing the neural stem or progenitor cell pool, adult neurogenesis, and gliogenesis by regulating the Stat3 and Notch signaling pathways, as well as through O-GlcNAc modification of Stat3 and the transmembrane and intracellular domains (TM/ICD) of Notch. These regulatory mechanisms play a critical role in shaping cognitive functions, underscoring the significance of O-GlcNAc in the maintenance and function of the adult nervous system [54, 55]. Utilizing chemoproteomics, close to a thousand proteins have been identified as subject to O-GlcNAcylation in mouse embryonic stem cells (ESCs). Notably, certain pluripotency transcription factors, such as Sox2, have been demonstrated to undergo functional regulation by O-GlcNAc [56]. The more detailed signaling mechanisms underlying O-GlcNAc-regulated NSCs/NPCs development remain further investigation.
The influence of O-GlcNAc extends beyond embryonic development, playing a crucial role throughout postnatal and adult stages. O-GlcNAc modification is essential for the survival and maintenance of neurons and astrocytes in mice. The loss of O-GlcNAcylation in forebrain excitatory neurons leads to progressive neurodegeneration, characterized by pathogenic processing of Tau and amyloid precursor protein (APP), neuronal death, gliosis, and memory impairment [57]. Conditional knockout of Ogt in sensory neurons leads to diminished behavioral sensitivity to mechanical and thermal stimuli, accompanied by reduced innervation of the epidermis and loss of cell bodies in the dorsal root ganglia [58]. In cultured hippocampal neurons, depletion of Ogt disrupts neuronal maturation via the Wnt/β-catenin signaling pathway, manifesting as reduced dendritic numbers and lengths, as well as immature spine development [59]. Furthermore, Ogt-mediated O-GlcNAc modification exerts a pivotal regulatory role in the proliferation of cerebellar granule cells, cerebellar development, and motor coordination [60, 61]. Ogt modulates the neurogenesis and neural differentiation of granule neuron precursors (GNPs) by activating the Sonic hedgehog (Shh) signaling pathway through O-GlcNAcylation at Ser355 of GLI family zinc finger 2 (Gli2) in mice. The O-GlcNAc modification of Gli2 induces its deacetylation and augments transcriptional activity via dissociation from p300, a histone acetyltransferase [60]. Meanwhile, G-protein subunit α12 (Gα12) undergoes Ogt-mediated O-GlcNAcylation, facilitating its interaction with Rho guanine nucleotide exchange factor 12 (Arhgef12) and subsequent activation of the RhoA/ROCK signaling pathway. Activation of RhoA/ROCK via the LPA pathway has been shown to mitigate developmental deficits observed in Ogt-deficient cerebellar granule cells [61]. Moreover, within the medial prefrontal cortex (mPFC), astrocytic Ogt exerts influence over the expression of proteins, with a considerable proportion of these altered proteins being implicated in metabolic processes, transferase activity, and biosynthetic pathways [62].
Notably, O-GlcNAc modification plays an essential role in the survival and maintenance of neurons, with pronounced expression observed in the cerebellum, particularly within Purkinje neurons. Recent studies have elucidated the critical role of O-GlcNAc modification for the viability of cerebellar Purkinje cells by inhibiting intracellular reactive oxygen species (ROS) production [63]. The inhibition and deletion of Ogt in rat cortical neurons and mouse Purkinje cells both trigger autophagy, a fundamental cellular process that involves the self-digestion of cytoplasmic components, culminating in an elevated expression of LC3-II [63, 64]. The regulation of O-GlcNAcylation through Ogt inhibition has been demonstrated to modulate mTOR-dependent autophagy. Moreover, the deletion of Ogt exerts a discernible impact on intracellular ROS generation, thereby influencing Purkinje cell viability [63]. Conversely, pharmacological inhibition of Oga causes accumulation of protein O-GlcNAc modification, which in turn inhibits proteasome activity and induces neuronal apoptosis in the brain [65].
The role of O-GlcNAc in synaptogenesis and synaptic plasticity
The formation, maturation, and transmission of synapses constitute the foundation for inter-neuronal signaling. Neuronal cells-specific conditional knockout and pharmacological inhibition of Oga or Ogt have been utilized to modulate O-GlcNAc levels to investigate the role of O-GlcNAc in cultured neurons and brain slices [32, 34, 66,67,68]. In primary motoneurons derived from heterogenous nuclear ribonucleoprotein (Hnrnpr) knockout mice, axon growth is reduced, accompanied by decreased synthesis of cytoskeletal and synaptic components [69]. These mutant mice exhibit denervated neuromuscular junctions and impaired motor function. Within axons, hnRNP R is part of translation initiation complexes and interacts with Ogt to modulate the O-GlcNAcylation of eIF4G [69]. In hippocampal slices, application of Oga/Ogt inhibitors showed no discernible effect on basal synaptic transmission in the CA3 to CA1 pathway [32, 66]. However, modulation of O-GlcNAc levels by pharmacological inhibitors showed inconsistent readouts regarding synaptic plasticity assay. Augmentation of O-GlcNAcylation via Oga inhibitors, such as Thiamet-G (TMG) or PUGNAc, has been found to exert a negative modulation on synaptic plasticity, enhancing long-term potentiation (LTP) while attenuating long-term depression (LTD) in the hippocampal CA3-CA1 pathway [67]. Studies employing knockout mouse models seem to corroborate these findings, demonstrating a dampening effect on synaptic plasticity. Specifically, LTP and LTD were diminished in the CA3 to CA1 pathway of hippocampus in Oga heterozygotes mice [68], whereas LTP was augmented in the CA3 to CA2 pathway of forebrain-specific Ogt conditional knockout mice [34]. In principal cells derived from rodent hippocampus, an acute elevation in O-GlcNAcylation was observed to attenuate GABAergic currents and reduce synaptic spikes through synaptic depression and reduced intrinsic excitability [70].
Ogt appears to regulate excitatory synaptic function, as evidenced by studies utilizing primary neuronal cell cultures wherein Ogt was genetically deleted either broadly or sparsely in neurons. Depletion of Ogt in vitro resulted in a reduction in mature morphological synapses and dendritic spines, with the spines present on Ogt knockout neurons predominantly exhibiting an immature phenotype [31]. While acute inhibition of Oga or Ogt exerts immediate suppressive effects, the cumulative impact of Oga gene deletion in conventional knockout mice is chronic. Additionally, the use of conventional knockout mouse models raises concerns regarding developmental effects. Although manipulation of Oga and Ogt provides insight into their significance in synaptic transmission, how O-GlcNAcylation of specific proteins influences synaptic transmission needs to be further investigated. Within hippocampal axons, high extracellular glucose levels coupled with Ogt activation have been demonstrated to attenuate mitochondrial motility. Notably, Milton, a mitochondrial motor-adaptor protein, serves as a requisite substrate for Ogt-mediated suppression of mitochondrial motility via O-GlcNAcylation at critical serine residues, including S447, S829/30, and S938 [71]. Proper circuit function in the nervous system necessitates inhibitory GABAergic transmission. In Caenorhabditis elegans, Ogt is highly enriched within presynaptic terminals of GABAergic motor neurons, where it combines with the ubiquitin ligase Eel-1 to form the Ogt/Eel-1 complex. This complex is essential for regulating synapse formation and modulating their sensitivity to aldicarb in a cell-autonomous manner [72].
A previous study revealed that 19% of synaptosome proteins undergo O-GlcNAcylation [73]. The synaptic proteins identified to undergo O-GlcNAc modification, include Bassoon, Piccolo, Shank2, Synapsin I, Synaptopodin, guanylate kinase-associated protein (GKAP), and ankyrin G [28, 74,75,76,77]. Synapsin I, a presynaptic protein linked to synaptic vesicles, modulates the release of these vesicles by regulating their transition from the reserve pool (RP) to the readily releasable pool (RRP). Under normal conditions, Synapsin I aids in maintaining synaptic vesicles within the RRP. However, following an action potential, phosphorylated Synapsin dissociates from the synaptic vesicles, enabling their relocation to the RP and eventual release. While definitive proof is absent, it is hypothesized that O-GlcNacylation of Synapsin I at T87 residue may yield effects akin to phosphorylation [78]. In the rat brain, seven O-GlcNAcylation sites (Ser55, Thr56, Thr87, Ser516, Thr524, Thr562, and Ser576) in Synapsin I are clustered near its five phosphorylation sites, located in domains B and D. O-GlcNAcylation seems to have a more profound and direct role in Synapsin I interactions, going beyond mere modulation of its phosphorylation [79]. GluA2, a subunit of aminomethyl phosphonic acid receptors (AMPARs) crucial for synaptic transmission and plasticity, functions as a pivotal postsynaptic voltage-gated channel. While the precise site of O-GlcNAcylation on GluA2 remains elusive, previous studies suggest that GluA2 O-GlcNAcylation may facilitate its internalization. Augmented global O-GlcNAcylation levels induced by an Oga inhibitor elicited a distinct form of LTD predicated on GluA2 internalization [67, 80]. Conversely, the Ogt inhibitor treatment resulted in elevated GluA2 surface expression [32]. Ogt assumes a critical role in the establishment of excitatory synapses, in part by selectively modulating the synaptic abundance of GluA2 and GluA3 subunits, pivotal constituents of AMPARs [31]. Consequently, given the absence of comprehensive elucidation regarding the specific site of O-GlcNAcylation on GluA2, further studies are required to investigate the effects of O-GlcNAcylated GluA2 on synaptic transmission.
Physiological functions of O-GlcNAcylation in the brain
The coordination of thousands of proteins by only two enzymes, OGT and OGA through O-GlcNAc modification, has recently begun to be explored in depth. The O-GlcNAcylation of these proteins plays a crucial regulatory role in the normal physiological functions of cells within the CNS. Dysregulation of O-GlcNAcylation can lead to various diseases, highlighting its critical role in cellular homeostasis. O-GlcNAc serves as a nutrient sensor in the brain, with the cellular identity and the extensive morphology of neurons and glia determining how O-GlcNAc responds to both general and neuron-specific stimuli, as well as the functional consequences of these responses. The specific neuronal circuits that drive behavior provide the context for understanding the role of O-GlcNAc. Hereby, we summarized the major physiological functions of O-GlcNAcylation in the brain, emphasizing its multifaceted impacts on neural functions and related diseases.
The role of O-GlcNAc in learning and memory
In mice, elevated levels of O-GlcNAc have been demonstrated to impede spatial learning and memory [68]. Additionally, hyper-O-GlcNAcylation has been implicated in disrupting hippocampal synaptic plasticity and causing dysregulation of AMPARs, thereby impairing LTP and LTD. Consistent with Oga deletion, pharmacological inhibition of Oga, which induces acute elevation of O-GlcNAcylation, has been shown to attenuate hippocampal activity by diminishing the intrinsic excitability of CA1 neurons and excitatory synaptic transmission at Schaffer collateral-CA1 synapses [80]. Furthermore, dynamic O-GlcNAcylation of cyclic AMP-response element-binding protein (CREB) has been demonstrated to enhance long-term memory consolidation [73]. The transcription factor CREB assumes a pivotal role in orchestrating various neuronal processes. In primary cultured neurons, neuronal activity triggers calcium- and kinase-dependent O-GlcNAcylation of CREB at Ser40. This modification at Ser40 exerts a suppressive effect on CREB activity, dendritic and axonal growth, while overexpression of S40A mutant within the amygdala has been shown to enhance long-term memory consolidation [73].
Targeting dynamic O-GlcNAcylation presents a promising avenue for regulating cognitive function in aging mouse brains. Indeed, neuronal Ogt overexpression and augmented O-GlcNAc levels in the aged hippocampus have been demonstrated to partially ameliorate age-related deficits in spatial learning and memory, along with associative fear memory in aged mice [34]. Furthermore, within the mesolimbic dopamine (DA) system, O-GlcNAcylation in ventral tegmental dopaminergic neurons is implicated in regulating motor learning and reaction to natural rewards. Ogt overexpression in ventral tegmental area (VTA) DAergic neurons leads to learning impairments in operant responses to sucrose and deficits in motor learning in the rotarod test [81].
Additionally, deletion or mutation of Oga in Drosophila results in deficits in habituation, a form of learning that is evolutionarily conserved [82]. Additionally, deficiency in O-GlcNAcase affects the quantity of synaptic boutons located at the axon terminals of neuromuscular junctions in larvae. These findings underscore the role of Oga in synaptic morphology and cognition in Drosophila, highlighting the significance of O-GlcNAc for cognitive function across different species.
The role of O-GlcNAc in brain energy metabolism
O-GlcNAc signaling represents a pivotal glycosylation modification that acts as a cellular sensor for nutrient status, exerting significant influence over various physiological processes, particularly in energy metabolism. Dysregulation of O-GlcNAc modification has been associated with a range of nutrition-related disorders, encompassing obesity, diabetes, cardiovascular diseases, neoplastic conditions, and neurological pathologies [40, 41, 46, 50, 83,84,85]. In cortical and hippocampal neurons, neuronal activity enhances mitochondrial O-GlcNAcylation, a process driven by glucose consumption during heightened activity, which allows neurons to adapt their energy expenditure based on fuel availability [86]. Disruption of O-GlcNAcylation dynamics impairs the ability of neurons to meet increased metabolic demands. Elevated activity of Ogt leads to dynamic O-GlcNAcylation of the regulatory domain of hexokinase 1 (HK1), facilitating the formation of the glycolytic metabolon on the outer mitochondrial membrane. This modification increases the association between mitochondria and HK1, facilitating coordinated glycolytic and mitochondrial ATP production [87]. In murine models, targeted deletion of Oga within the brain during embryonic stages yields notable pleiotropic phenotypes, such as growth defects, early-onset obesity, and metabolic dysregulation persisting into adulthood [88]. Notably, fasting triggers heightened O-GlcNAc levels within the ventromedial hypothalamus (VMH), while specific Ogt knockout within VMH neurons precipitates rapid weight gain, adiposity, and reduced basal energy expenditure in normally nourished mice [89]. Conditional knockout of Ogt via αCaMKII-CreERT2 in the adult murine brain, inclusive of the hypothalamus, has been correlated with transient obesity attributed to augmented food intake and duration [90, 91]. This phenomenon has been attributed to reduced excitability in a subset of αCaMKII-positive and Ogt-deficient neurons within the paraventricular nucleus (PVN) [91]. Furthermore, Ogt knockout in inducible αCaMKII-CreERT2-expressing neurons resulted in notable neuronal depletion across diverse brain regions, particularly manifesting reduced hypothalamic neuronal populations expressing the leptin receptor, notably within the arcuate nucleus [90]. Perturbation of Ogt function within the VMH impairs neuronal excitability, diminishes sympathetic outflow, and disrupts sympathetic innervation of white adipose tissue. Such dysregulation during developmental stages can lead to enduring metabolic derangements extending into adulthood.
The hypothalamus regulates essential bodily functions, such as food intake, body temperature, blood circulation, the sleep-wake cycle, fluid and electrolyte balance, metabolism and sexual behavior [92, 93]. O-GlcNAc assumes a critical role in governing hypothalamic glucose and lipid homeostasis. In the arcuate nucleus of hypothalamus, two distinct neuronal populations exist: orexigenic neurons expressing hypothalamic agouti-related peptide/neuropeptide Y (AgRP/NPY) and anorexigenic neurons expressing proopiomelanocortin (POMC), which play integral roles in responding to peripheral hormones and nutrients, thereby maintaining energy homeostasis and regulating glucose metabolism [94]. Notably, the expression of Ogt exhibits pronounced elevation in hypothalamic AgRP neurons, an effect that is further enhanced by fasting and ghrelin stimulation. Abatement of O-GlcNAc levels consequent to Ogt ablation within AgRP neurons has been demonstrated to elicit white adipose tissue browning. Moreover, O-GlcNAc intricately modulates hypothalamic hunger signaling by impinging upon the excitability and responsiveness of AgRP neurons to ghrelin. Conversely, fasting and ghrelin stimulation have been shown to heighten Ogt expression and O-GlcNAc modification levels in AgRP neurons within the arcuate nucleus [33]. Specifically, Ogt exerts regulatory effects on the Kv7.3 potassium channel by specifically targeting Thr655 residue, thereby modulating AgRP neuronal firing and influencing overall neuronal activity. Importantly, this potassium channel is also implicated in the suppression of white fat browning and the prevention of diet-induced obesity [33]. Furthermore, O-GlcNAc is implicated in the orchestration of hypothalamic satiety processing [95]. In sum, O-GlcNAc signaling in the brain assumes a pivotal role in modulating glucose and lipid metabolism in animals by influencing hormone secretion, neuronal excitability, and neuronal viability in the hypothalamus.
In C. elegans, loss of Ogt function correlates with heightened regenerative outgrowth via a defined metabolic pathway [96]. Glycolysis, one-carbon metabolism (OCM), the serine synthesis pathway (SSP), and the downstream transsulfuration metabolic pathway (TSP) all emerge as integral contributors to the augmented regenerative capacity observed in Ogt-deficient neurons. Importantly, the regenerative phenotype associated with Ogt mutation is rescinded upon disruption of OCM and the SSP, thus genetically and pharmacologically linking OCM to glycolysis [96]. Insights gleaned from model organisms such as C. elegans offer valuable elucidation into the broader ramifications of O-GlcNAc cycling. In Drosophila, excessive dietary sugar intake leads to gustatory deficits and hyperphagia through the cell-autonomous action of Ogt within sweet-sensing neurons. Manipulation of sweet gustatory neuron excitability or modulation of Ogt expression confers protection against diet-induced adiposity in fruit flies [97]. Collectively, evidence underscores the evolutionary conservation of O-GlcNAc’s role in neural regulation of metabolism across diverse species.
The role of O-GlcNAc in circadian rhythms regulation
Circadian rhythms are meticulously regulated through a complex network of transcriptional and translational feedback loops. In adult Zebrafish, disruption of the circadian rhythm impairs the rhythmic fluctuations of O-GlcNAcylation and alters the expression patterns of Ogt and Oga in the brain, significantly hindering learning and memory functions. And pharmacological inhibition of Ogt using OSMI-1 has been shown to disrupt the wake-sleep patterns [98]. In the context of Drosophila, O-GlcNAcylation has been identified as a critical PTM that governs circadian regulation and influences the pace of the internal clock [99]. The PERIOD protein (Per), a key factor in regulating circadian clock speed in Drosophila, is directly targeted by Ogt. The level of O-GlcNAcylation in Per undergoes daily fluctuations, mainly during the initial half of the night, a period when Per is primarily localized in the cytoplasm. Notably, the timing of Per’s nuclear translocation is either advanced or delayed depending on Ogt levels in Drosophila. Experimental manipulation of Ogt levels within the clock neurons of Drosophila has shown the ability to modulate the duration of circadian behavioral rhythms [99]. Through mass spectrometry-based proteomics, Serine 942 (Ser942) of Per was recognized as a critical O-GlcNAcylation site. Functional in vivo characterization of this modification revealed that O-GlcNAcylation at Per Ser942 weakens the interaction between Per and Clock, a pivotal transcriptional activator of clock-controlled genes. This reduced interaction is thought to delay the initiation of the circadian repression phase, thereby preventing premature repression, and ensuring proper rhythmicity [100].
In addition to its role in modifying Per, O-GlcNAcylation has been shown to influence other circadian rhythm-related proteins. In the murine heart, the clock component Bmal1 undergoes O-GlcNAcylation. Pharmacological inhibition of Oga, which increases global O-GlcNAcylation levels, leads to reduced Per2 protein levels, time-of-day-dependent expression of Bmal1, and advancements in the phase of the suprachiasmatic nucleus (SCN) clock [101]. Additionally, in both murine livers and U2OS cells, the expression of Ogt transcripts oscillates in a circadian manner, with Bmal1 and Clock as key targets. The rhythmic O-GlcNAcylation of these proteins inhibits their ubiquitination, thereby stabilizing Bmal1 and Clock and promoting the activation of Clock-controlled genes [102]. Moreover, O-GlcNAcylation of Bmal1 in hippocampal neurons of diabetic mice has been demonstrated to enhance its interaction with Clock, resulting in dysregulated circadian clock gene expression. This aberrant regulation induces mitochondrial calcium overload, ultimately leading to neuronal damage and contributing to cognitive decline [103].
Ogt also plays a pivotal role in modulating the phosphorylation status and activity of its target clock proteins by interacting with kinases and competing with phosphorylation via O-GlcNAcylation [104]. Notably, Ogt itself is a substrate of glycogen synthase kinase 3β (GSK3β), a serine/threonine kinase, with which it shares a reciprocal regulatory relationship. In the context of circadian rhythm regulation, core clock proteins such as Clock and Per2 are subject to reversible O-GlcNAcylation, a modification that modulates their transcriptional activities. Furthermore, the competitive interaction between O-GlcNAcylation and phosphorylation at the Per2 S662–S674 region—a site crucial for regulating the human sleep phase—has been identified. This interaction is at least partially influenced by glucose levels, suggesting a metabolic link in the modulation of circadian function [104]. Collectively, these studies offer compelling evidence for the involvement of O-GlcNAcylation in circadian regulation. However, further investigation is needed to achieve a comprehensive understanding of the neural and molecular mechanisms underlying its impact on circadian physiological regulation in the brain.
The role of O-GlcNAc in other physiological functions
Stress is crucial in maintaining homeostasis and ensuring the normal physiological activities of the body. The preservation of mitochondrial integrity and the mitigation of cellular stress is vital for the fundamental physiology and survival of neurons. O-GlcNAcylation also plays an essential role in the regulation of stress process both in vitro and in vivo. Deletion of Ogt in NSCs induces ER stress in mice [30]. Similarly, in SH-SY5Y neuroblastoma cells and murine subjects, alterations in OGT expression or treatment with TMG exert notable effects on the expression of activating transcription factor 4 (ATF4), an important transcription factor involved in the mitochondrial integrated stress response [105].
In spinal motor neurons, the levels of O-GlcNAcylation increase in response to age-dependent oxidative stress. Pharmacological suppression of Oga has been demonstrated to mitigate the loss of spinal motor neuron in aged NPGPx-deficient mice, a phenotype characterized by heightened sensitivity to oxidative stress [106]. Furthermore, glucosamine treatment has been found to confer protection to in vitro retinal ganglion cells (RGCs) against oxidative stress-induced damage [107]. Recent studies by Fan et al. [36] have elucidated the implication of O-GlcNAc in astroglia from the medial prefrontal cortex (mPFC) in the development of depression after chronic social-defeat stress (CSDS) [36]. External stimuli, such as hypo-osmolarity-induced hyperosmotic stress have been shown to downregulate OGT expression and diminish O-GlcNAc levels. Interventions aimed at reducing cellular O-GlcNAcylation have been shown to facilitate the induction of the p75 neurotrophin receptor (p75NTR) by hypo-osmolarity, thereby establishing a direct link between protein O-GlcNAcylation, p75NTR induction, and hyperosmotic stress [108]. Therefore, the O-GlcNAcylation plays an important role in the regulation of stress response.
The role of O-GlcNAcylation in immune responses within the brain is a subject of ongoing investigation. Recent studies have elucidated mechanisms through which O-GlcNAcylation influences the neuroinflammation processes in neuronal cells. For instance, in BV2 mouse microglial cell line, lipopolysaccharides (LPS) exposure elevates the O-GlcNAcylation level of c-Rel and further promotes its binding to the NF-κB site within the iNOS promoter. Knockdown of Ogt reduces the O-GlcNAc level of c-Rel and impairs the interaction between c-Rel and p50 in response to LPS stimulation, but does not influence either c-Rel’s binding to the iNOS promoter or its transcriptional activity, whereas glucosamine (GlcN) treatment attenuates these LPS-induced effects, thereby exhibiting anti-inflammatory properties [109]. Furthermore, Oga inhibitor TMG treatment improves neurological deficits and clinical outcomes in neurobehavioral tests by regulating the expression of both pro-inflammatory and anti-inflammatory cytokines in an experimental stroke mouse model [110]. In astrocytes, Ogt deficiency triggers inflammatory activation and impairs cognitive function in mice. Enhancement of O-GlcNAcylation through O-GlcNAc supplementation suppresses astrocyte activation, thereby reduces inflammation, and enhances cognitive function in Ogt-knockout mice [35]. Notably, Ogt interacts with the p65 subunit of NF-κB and catalyzes the O-GlcNAcylation of p65 at serine 384 (S384). This PTM impedes the activation of the NF-κB signaling pathway by preventing the binding of glycogen synthase kinase 3 beta (Gsk3β) [35]. These results collectively suggest that O-GlcNAc modification plays an essential role in mediating neuronal stress responses and promoting neuroprotection following injury. The modulation of O-GlcNAcylation represents a promising therapeutic strategy for managing neuroinflammatory conditions to enhance neuronal resilience.
Together, O-GlcNAcylation functions as a critical regulator in diverse facets of neurobiology, encompassing neural development, cognition, metabolism, circadian rhythms and stress response. It exerts significant influence over embryonic and adult neurogenesis, neuronal apoptosis, memory consolidation, and metabolic pathways (see Fig. 1). Meanwhile, Within the CNS, research efforts have been primarily focused on elucidating the precise molecular mechanisms that underpin O-GlcNAcylation in neurons and other cellular subsets. Comprehensive proteomics analyses have identified a wide array of proteins that undergo O-GlcNAcylation in neuronal contexts, particularly within nuclear and synaptic compartments. And some O-GlcNAcylated proteins and the downstream signaling pathways in NSCs, NPCs, neurons, astrocyte and microglia have been identified and elucidated, respectively (see Fig. 2). However, the elucidation of the specific functions attributable to O-GlcNAcylated proteins in neuronal physiology remains somewhat limited (see Table 1). This limitation is partly due to the scarcity of site-specific O-GlcNAc antibodies and methodologies that facilitate site-directed mutagenesis to mimic O-GlcNAc-deficient protein states, in contrast to the well-established resources available for phosphorylation-centric studies. Future studies should aim to overcome the current methodological limitations to acquire a deeper understanding of the functional implications of O-GlcNAcylation in neuronal physiology and its role in disease pathogenesis.
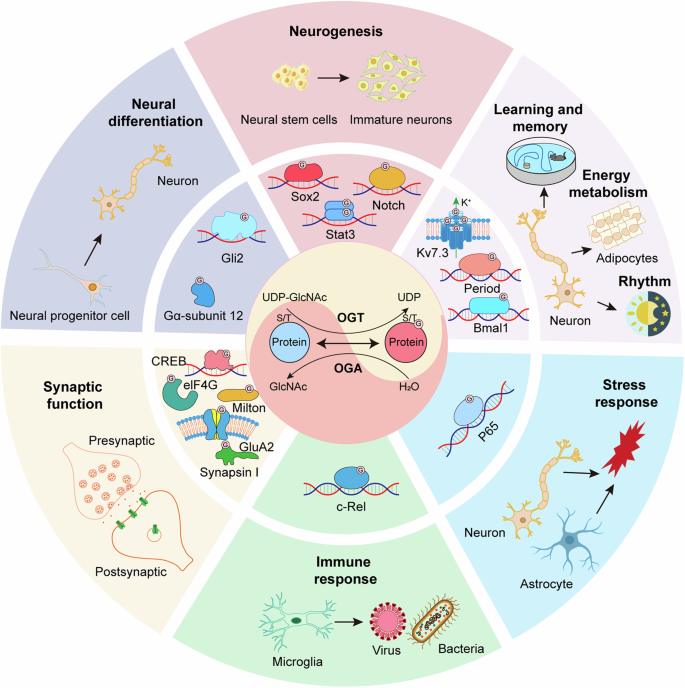
The nutrient flux through the hexosamine biosynthetic pathway (HBP) generates UDP-GlcNAc, which undergoes dynamic cycling onto proteins as O-GlcNAcylation by OGT and OGA. O-GlcNAc modification in neural stem cells (NSCs), neural progenitor cells, neurons and glia regulate diverse neurodevelopmental and physiological functions, including neural development, synaptic function, learning and memory, metabolism, stress response, rhythm, and immune response.

O-GlcNAcylation in neural stem cell (NSC), neural progenitor cell, neuron, astrocyte, and microglia target diverse proteins and downstream signaling pathway to regulate diverse neurodevelopmental and physiological functions.
Pathological roles of O-GlcNAcylation in brain disorders
O-GlcNAcylation has been established as a crucial process in mammals, and its dysregulation has been implicated in the pathological mechanisms underlying a wide array of human diseases, such as cancer [83, 84], type II diabetes [16], cardiac hypertrophy [85, 111], and CNS diseases and injuries [40, 41, 44, 46, 47, 49, 50]. In neurodevelopmental disorders, aberrant O-GlcNAcylation has been associated with structural and functional anomalies, such as abnormal cell proliferation, impaired neuronal differentiation, and premature neurodegeneration. Clinical evidence further underscores the connection between O-GlcNAcylation and conditions like intellectual disability (ID), Rett syndrome (RTT), and Down syndrome (DS). The O-GlcNAc pathway also plays a role in neurodegenerative diseases, with age-related alterations in O-GlcNAcylation levels impacting cognitive function and contributing to the progression of conditions such as Alzheimer’s disease (AD). Additionally, O-GlcNAcylation is implicated in neuropsychiatric disorders, including schizophrenia (SZ) and major depressive disorder (MDD), though its exact role in these conditions is still under investigation. Moreover, O-GlcNAcylation is involved in CNS injuries, such as ischemic stroke, intracerebral hemorrhage (ICH), subarachnoid hemorrhage (SAH), and traumatic brain injury (TBI), where its modulation holds potential therapeutic promise. Overall, elucidating the role of O-GlcNAcylation in these conditions is crucial for developing effective interventions and enhancing brain health throughout the lifespan. In this section, we have summarized the intricate connections between O-GlcNAcylation and the pathogenesis of these brain disorders (see Fig. 3).

Dysregulation of O-GlcNAcylation is associated with neurodevelopmental diseases, psychiatric diseases, and neurodegenerative diseases, as well as CNS injuries. CNS, central nervous system; SAH, subarachnoid hemorrhage; ICH, intracerebral hemorrhage; TBI, traumatic brain injury; XLID, X-linked intellectual disability; RTT, Rett syndrome; DS, Down syndrome; SZ, Schizophrenia; MDD, major depressive disorder; AD, Alzheimer’s disease; PD, Parkinson’s disease; HD, Huntington’s disease; ALS, Amyotrophic lateral sclerosis; MS, Multiple sclerosis. The downward arrow (in blue) indicates that a reduction in O-GlcNAcylation is linked to the occurrence of the disease, or that upregulation of O-GlcNAcylation is protective. The upward arrow (in red) indicates that an elevation of O-GlcNAcylation is correlated with the disease, or that suppression of O-GlcNAcylation is protective.
O-GlcNAcylation in neurodevelopmental diseases
The ablation of either OGT or OGA specifically in neurons led to profound structural and functional anomalies, stemming from neurodevelopmental defects or premature neurodegeneration in animal models [57, 88, 112]. For instance, neuron-specific Ogt deficient mice led to abnormal development and early postnatal lethality, attributed to the accumulation of hyperphosphorylated Tau protein [113]. Moreover, Ogt deficiency in NSCs resulted in a range of severe brain developmental impairments, such as decrease in cell proliferation, inhibition of neuronal dendritic and axonal differentiation, shrinkage of cortex and hippocampus, and apoptosis of neurons [30]. In addition, knockout of Oga in the mouse brain caused abnormal cell proliferation and developmental delay, manifesting as delayed brain neurogenesis and differentiation [88]. It has been reported that O-GlcNAcylation regulates various transcription factors that play crucial roles in stem cell pluripotency and early neuronal differentiation, such as OCT4, SOX2, NANOG, SOX1, and OTX2 [88, 114,115,116]. Collectively, these findings suggest that dysfunction in O-GlcNAcylation may serve as an important contributor to neurodevelopmental diseases in animals.
Recently, increasing clinical evidence underscores the connection between O-GlcNAcylation and neurodevelopmental diseases, such as ID [37, 38, 40], RTT [117], and DS [118]. Notably, mutations in the OGT gene have been identified in patients with ID, and genetic analysis strongly suggest a link between these mutations and the disease [37, 38, 40]. Interestingly, the dysregulation of O-GlcNAcylation cause phenotypes associated with ID in both mouse and Drosophila models, including microcephaly and deficits in locomotor function and learning [119, 120]. The etiology of ID is complex, encompassing both genetic and non-genetic factors [121, 122]. Intriguingly, ID is more prevalent among males and is associated with over 100 ID-related genes located on the X chromosome [123,124,125,126]. The preferential expression of X-linked genes in the brain compared to other tissue [127] underscores their crucial role in brain function.
Notably, the OGT gene resides on the X chromosome in both humans and mice, and mutations in this gene have been detected in patients with X-linked intellectual disability (XLID) [37, 38]. Several pathogenic variants of OGT have been reported in XLID patients [39, 40], all leading to delayed development, brain abnormalities, and reduced intellectual capacity, further strengthening the connection between the O-GlcNAc system and neurodevelopment. Mutations in the X-linked methyl-CpG-binding protein 2 (MECP2) gene are the primary cause of RTT, a progressive neurodevelopmental disorder linked to the X chromosome. Interestingly, MECP2 undergoes dynamic O-GlcNAc modification at threonine 203 (T203), and disruption of O-GlcNAcylation at this specific site in MECP2 impairs dendritic development and dendritic spine maturation in cultured hippocampal neurons, and dysfunction in synaptic transmission in the mouse cerebral cortex [117]. Individuals with DS are at a high risk for the early development of AD pathology. Studies have shown that disruptions in O-GlcNAcylation linked to the appearance of AD hallmarks in the brain of DS mouse model, and rescue of O-GlcNAcylation homeostasis could reduce AD-related hallmarks [118]. Given the intricate links between O-GlcNAcylation and neurodevelopmental diseases, further investigation into the role of this modification in these disorders is crucial for understanding their pathogenesis and developing effective therapeutic strategies.
O-GlcNAcylation may serve as a critical mediator between nutritional impairment and brain developmental defects. Since O-GlcNAcylation depends on uridine diphosphate-N-acetylglucosamine (UDP-GlcNAc), a product derived from glucose, diet is pivotal in modulating its activity. In turn, O-GlcNAcylation regulates signaling pathways via PTMs in reaction to fluctuations in blood glucose levels. Placental blood circulation is initiated as early as the end of the first trimester during embryonic development [128]. Multiple crucial factors, including food cravings, increased sugar intake and gestational diabetes, can affect a mothers’ circulating glucose levels, which subsequently influence the O-GlcNAcylation levels in the developing fetus [129]. Consequently, O-GlcNAcylation has been hypothesized to partly explain the impact of a mother’s diet on the development of her offspring [130, 131]. Growing evidence indicates a correlation between altered glucose levels, O-GlcNAcylation, and brain developmental defects [88, 132]. Notably, glucose levels have been linked to delayed brain development in diabetic patients [133]. Disruption of placental Ogt can affect the Hypothalamus-Pituitary-Adrenal (HPA) axis, crucial for fetal growth and metabolism, thereby establishing a link between placental Ogt and neurodevelopmental programming in mice [134].
O-GlcNAcylation in neurodegenerative diseases
O-GlcNAcylation is a critical PTM that plays a pivotal role in modulating synaptic and neuronal functions. It is reasonable to observe altered O-GlcNAc levels during aging, as these changes can significantly compromise neural functions in the brain. Accumulating evidence indicates that the level of O-GlcNAcylation varies with age [54, 135]. For instance, both the expression of Ogt and the level of O-GlcNAcylation are lower in the hippocampus of aged mice compared to young mice, resulting in impaired cognitive function in aged animals, and enhancing neuronal O-GlcNAcylation could potentially improve cognitive function [34]. Similarly, age-related decreases in O-GlcNAcylation in NSCs promote a glial fate switch in the hippocampus, further contributing to age-related neurodegenerative changes [54].
Numerous proteins associated with aging-related diseases, such as microtubule-associated protein Tau [41], β-Amyloid precursor protein (APP) [42], α-Synuclein (α-syn) [136,137,138], are O-GlcNAcylated. Alteration in their O-GlcNAcylation status have been extensively linked to neurodegeneration [43,44,45, 139], highlighting the importance of O-GlcNAcylation in maintaining neural health throughout the lifespan.
O-GlcNAcylation and Alzheimer’s disease
AD ranks among the most common neurodegenerative disorders, featured by the accumulation of amyloid plaques and neurofibrillary tangles (NFTs). Numerous studies have indicated that brain glucose uptake and metabolism are compromised in AD patients, with O-GlcNAc levels being reduced by 22 to 50% compared to healthy individuals [41, 57, 140]. This metabolic dysregulation can be detected at an early stage, preceding the emergence of external clinical symptoms, and is closely associated with the severity of cognition decline. This suggests that reduced O-GlcNAcylation may play a pivotal role in the development of AD [140].
Furthermore, two key proteins, Tau and APP, have been reported to undergo extensive O-GlcNAcylation. Higher levels of O-GlcNAcylation have been demonstrated to protect against the toxic effects of Tau and amyloid-β peptide [41, 42]. Additionally, two primary brain glucose transporters, GLUT1 and GLUT3, which facilitate glucose uptake into neurons, are found to be downregulated in the brains of AD patients. This downregulation is linked to reduced O-GlcNAcylation, hyperphosphorylation of Tau, and increased density of NFTs [141]. Moreover, a significant reduction in the levels of O-GlcNAcylated clathrin assembly protein-3 (AP-3) has also been observed in AD [142], indicating a potential association between O-GlcNAcylated AP-3 and the pathological mechanisms underlying AD.
O-GlcNAcylation and Parkinson’s disease
Parkinson’s disease (PD) is arising from the gradual loss of midbrain neurons responsible for releasing dopamine. Lee and colleagues have demonstrated that O-GlcNAcylation is essential for the survival and function of dopamine neurons. A downregulation of O-GlcNAcylation in these neurons results in severe motor defects, while an upregulation mitigates neurodegeneration, synaptic dysfunction, and motor deficits [112]. This underscores the critical role of O-GlcNAcylation in PD pathology.
A defining neuropathological characteristic of PD is the accumulation of insoluble proteinaceous inclusions [143], known as Lewy bodies (LBs) in cell bodies and Lewy neurites (LNs) in neurites. The degeneration of dopamine neurons in PD is often accompanied by the aggregation of α-syn and other proteins into LBs, leading to synaptic impairments and progressive neuronal death in PD brains [144,145,146]. Notably, in addition to PD, the accumulation of α-syn in insoluble aggregates has also been identified in multiple system atrophy (MSA) and dementia with Lewy bodies (DLB) [147,148,149]. Convincing evidence indicates that α-syn undergoes O-GlcNAc modification at multiple residues in vivo [136, 138]. This modification reduces its aggregation propensity and associated pathology [136,137,138, 150]. Furthermore, O-GlcNAcylation inhibits the proteolytic cleavage of α-syn by calpain [151], indirectly suppressing its aggregation. Pharmacological inhibition or genetic knockdown of OGA inhibits the internalization of α-synuclein fibrils [152], thereby limiting the spread of pathology throughout the brain.
O-GlcNAcylation and Huntington’s disease
Huntington’s disease (HD) is a rare, hereditary neurodegenerative disorder featured by progressive neurodegeneration, leading to progressive motor dysfunction and cognitive decline. It is mainly due to a mutation in the huntingtin (HTT) gene, which codes for the huntingtin protein (HTT). Both HTT and huntingtin-interacting protein 1-related protein (HIP1R) appear to undergo O-GlcNAcylation [112, 153], indicating a possible role for O-GlcNAcylation in HD. Nucleoporin (NUP), a constituent of the nuclear pore complex, has been found to be associated with HD pathology in HD mouse models and postmortem brains of HD patients [154]. Intriguingly, NUP undergoes extensive O-GlcNAcylation, which is critical for maintaining the structural stability of nuclear pores and their ability for selective filtration [155]. However, the particular role and mechanisms of O-GlcNAc in the pathogenesis of HD remains unclear.
It has been reported that suppression of O-GlcNAcylation increases basal autophagy flux and enhances the clearance of toxic protein aggregates by improving autophagosome-lysosome function, thereby protecting against huntingtin aggregation and cytotoxicity mediated by mutant HTT (mHTT) in neuroblastoma cells and Drosophila models [156]. Therefore, while increasing evidence suggests a connection between O-GlcNAcylation and HD pathology, the exact function of O-GlcNAcylation within the context of HD pathology requires further exploration.
O-GlcNAcylation and amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS), a progressive neurological disorder affecting motor neurons, is strongly influenced by aging as a risk factor. Multiple factors play a role in the pathogenesis of ALS, including genetic mutations, oxidative stress, and inflammation [157]. Genetic mutation in SOD1 (superoxide dismutase 1), FUS (fused in sarcoma/translated in liposarcoma), TDP-43 (TAR DNA-binding protein 43), and C9orf72 (chromosome 9 open reading frame 72) are most frequently observed in ALS patients [158].
Increasing evidence indicates that O-GlcNAcylation is implicated in the occurrence of ALS. It was reported that O-GlcNAcylation levels in motor neurons of the spinal cord were dramatically reduced in a mutant SOD1-overexpressing ALS mouse model compared to controls [159]. Additionally, hyperphosphorylation and aggregation of neurofilaments (NFs) are observed in ALS pathology [160], and NFs are known to be modified by O-GlcNAc [161]. Consistent with these observations, there was a significant reduction in the O-GlcNAcylation of NFs in the mutant SOD1-overexpressing ALS rat model. These findings indicate an association between the disturbance of O-GlcNAcylation and ALS pathology [162], suggesting that O-GlcNAcylation may improve ALS pathology by suppressing hyperphosphorylation.
Furthermore, oxidative stress is another established cause of ALS. Nonselenocysteine-containing phospholipid hydroperoxide glutathione peroxidase (NPGPx) serves as an oxidative stress sensor and signaling molecule that modulates the antioxidative activity of its target proteins by shuffling disulfide bonds [163]. Studies suggest that ALS patients exhibit lower expression levels of NPGPx compared to unaffected individuals, and mice deficient in NPGPx develop ALS-like phenotypes. Further research suggests that NPGPx regulates O-GlcNAcylation by inhibiting Oga, thereby protecting motor neurons from degeneration [106]. Additionally, pathological TDP-43 aggregation is a hallmark of several neurodegenerative diseases, including frontotemporal lobar degeneration (FTLD) and ALS [164]. It has been demonstrated that TDP-43 is O-GlcNAcylated by OGT in vivo, and OGT-mediated O-GlcNAcylation of TDP-43 inhibits disease-related proteinopathies [165]. These findings indicate that O-GlcNAcylation can modulate ALS pathology and serve as a potential therapeutic target for ALS.
O-GlcNAcylation and multiple sclerosis
As previously mentioned, the role of O-GlcNAcylation in inflammation and immune responses within the CNS has also been documented [166]. Several studies demonstrate that O-GlcNAc modification is implicated in the pathogenesis of autoimmune diseases [167, 168]. Multiple sclerosis (MS) is one of the most prevalent chronic inflammatory autoimmune disorders characterized by chronic demyelination and neurodegeneration of the CNS in young adults, and CD4 + T cell–mediated autoimmunity is pivotal in the pathogenesis of MS. Microarray screening of peripheral blood mononuclear cells (PBMCs) has shown that OGT mRNA expression is elevated in MS patients compared to healthy controls [169]. In addition, miR-15b negatively regulates Th17 differentiation and MS pathogenesis by targeting OGT, thereby influencing the transcriptional activity of the retinoic acid–related orphan receptor (RORγT) through O-GlcNAcylation of NF-kB [170]. These findings suggest that O-GlcNAc modification may play a significant role in MS pathology.
Together, these findings not only deepen our understanding of the role of O-GlcNAcylation in neurodegenerative diseases but also offer new insights into potential therapeutic strategies for these conditions.
The role of O-GlcNAcylation in neuropsychiatric disorders
An increasing number of recent studies have demonstrated that O-GlcNAcylation played a critical role in the development of neuropsychiatric disorders. Disrupted O-GlcNAc modification has been identified in SZ and MDD. However, the exact role of O-GlcNAcylation in neuropsychiatric disorders is still far from understanding.
As a serious mental illness, SZ affects 1% of the world’s population and is featured by positive symptoms (e.g., hallucinations, delusions), negative symptoms (e.g., avolition, reduced emotional expression), and cognitive impairment. It has been reported that OGT mRNA levels in dorsolateral prefrontal cortex (DLPFC) are 253% higher in SZ patients than healthy persons, and OGA protein levels in superior temporal gyrus (STG) correspondingly reduced in SZ patients [171]. Enhanced O-GlcNAcylation has been shown to result in diabetes and mitochondrial dysfunctions, which have been identified in SZ patients [47]. A sudden rise in O-GlcNAcylation reduces GABAergic currents in principal cells of the rodent hippocampus, thereby disrupting the excitation/inhibition balance, a characteristic feature observed in SZ [47]. These studies suggest a potential link between O-GlcNAcylation and SZ. Further investigation of O-GlcNAcylation could offer promising avenues for developing therapeutic strategies for SZ.
MDD is a severe and pervasive psychiatric condition, primarily characterized by dysregulated glutamatergic signaling within the prefrontal cortex (PFC). MDD has been linked to deregulated glucose metabolism [172], specifically manifesting in a reduction of glucose metabolism in the PFC [173, 174]. In a rat model of depression, Ogt-related mitochondrial motility exhibits sex-specific differences and is influenced by exercise, highlighting its role in depression [47]. Additionally, Oga heterozygous (Oga+/−) mice, which exhibit chronically elevated levels of O-GlcNAcylation, display antidepressant-like behaviors, and altered synaptic transmission in the medial prefrontal cortex (mPFC) [46].
Notably, MDD patients show elevated OGT mRNA levels, and Ogt expression in astroglia is upregulated in the mPFC of susceptible mice following chronic social-defeat stress (CSDS). Pharmacological inhibition or astrocyte-specific knockout of Ogt in the mPFC leads to antidepressant effects, whereas overexpression of astrocytic Ogt in the mPFC increases stress susceptibility. Further research indicates that the modulation of astrocytic Ogt in the mPFC on depressive-like behaviors occurs via the O-GlcNAcylation of GLT-1, a crucial glutamate transporter for maintaining glutamatergic homeostasis in the brain [36]. This observation implies that modulating the O-GlcNAcylation of specific proteins, such as GLT-1, may offer a promising therapeutic approach for MDD.
Collectively, the growing understanding of O-GlcNAcylation’s involvement in neuropsychiatric disorders, particularly SZ and MDD, offers significant insights into the signaling mechanisms driving these pathologies and underscores potential avenues for developing innovative therapeutic approaches.
The role of O‑GlcNAcylation in CNS injuries
O-GlcNAcylation is implicated in the progression of CNS injuries, such as ischemic stroke [50], SAH [48], ICH [49], and TBI [175]. Modulation of O-GlcNAcylation has been shown to mitigate brain damage, making it a promising therapeutic target for addressing CNS injuries.
Ischemic stroke is a prevalent and severe condition that predominantly affects the elderly population. Current treatment strategies primarily focus on acute thrombolysis and/or thrombectomy to restore blood flow. However, these treatments often fall short in terms of their effectiveness, prompting the need for more effective therapeutic approaches. The role of O-GlcNAcylation in stroke offers a potential strategy. Although O-GlcNAcylation has been associated with stroke, there is ongoing debate regarding whether its effect is protective or detrimental. Research has shown that O-GlcNAcylation initially increases transiently but then declines during cerebral ischemia in relevant models [50]. Additionally, a moderate elevation (less than threefold) of brain O-GlcNAcylation has been demonstrated to be protective, reducing infarct size and motor deficits while improving cerebral ischemia-reperfusion injury [50]. In an experimental mouse model of stroke, the administration of TMG reduced infarct size, ameliorated neurological impairments, and enhanced clinical outcomes through regulating the expression of pro-inflammatory and anti-inflammatory cytokines [110]. These observations strongly suggest that O-GlcNAcylation plays a neuroprotective role in ischemia and cerebral reperfusion.
However, some studies also point out the harmful impact of O-GlcNAcylation in stroke [176]. In hyperglycemic mice, TMG accelerated cerebral ischemic injury, facilitated hemorrhagic transformation, and increased apoptosis, while O-GlcNAcylation suppression through the use of 6-diazo-5-oxo-L-norleucine mitigated the cerebral injury associated with ischemic stroke [176]. Collectively, O-GlcNAcylation plays a critical role in ischemic stroke, and its ultimate effect may vary depending on a range of factors, such as diverse experimental models, environmental conditions, and stimulatory circumstances. Therefore, further research is needed to fully understand and harness the potential of O-GlcNAcylation in the treatment of ischemic stroke.
Moreover, O-GlcNAcylation has been associated not only with ischemic stroke but also with SAH [48] and ICH [49]. SAH triggers cellular and ER stress, which activates the unfolded protein response (UPR) in nerve cells. O-GlcNAcylation has been recognized as a key factor in the pathophysiology of SAH. Xbp1s, the ultimate product of UPR due to ER stress, can stimulate the expression of the key regulatory enzyme GFAT1 in the hexosamine pathway, thereby increasing the level of O-GlcNAcylation and providing protection on neural cells [48]. Furthermore, mitochondrial O-GlcNAc modification has been shown to enhance the therapeutic efficacy of transplanted mitochondria in a mouse model of stroke, while higher levels of O-GlcNAcylation in mitochondria correlating with improved clinical outcomes in SAH patients [177]. ICH constitutes about 10–40% of stroke cases, and the immune regulatory mechanisms within the CNS are crucial during the ICH process. Research has indicated that O-GlcNAcylation, through Oga inhibition, enhances neurological function and reduces brain edema in mice subjected to experimentally induced ICH, likely by decreasing brain-infiltrating immune cells and enhancing the integrity of the blood-brain barrier [49].
Recently, O-GlcNAcylation has been reported to play a role in TBI [175]. In the TBI model of adult zebrafish, the needle stab-induced injury resulted in reduced neuronal proliferation and neurobehavioral deficits, accompanied by decreased levels of OGT and increased levels of OGA. Treatment with glucosamine (GlcN) restored O-GlcNAc levels and improved neurobehavioral functions, while inhibition of the hexosamine biosynthetic pathway using 6-diazo-5-oxo-L-norleucine blocked this recovery [175]. These findings indicate that the O-GlcNAc pathway could represent a potential therapeutic target for TBI.
Therefore, the O-GlcNAc pathway is widely involved in CNS injuries including TBI. However, the exact role of O-GlcNAc in specific injuries needs further investigation, which may contribute to the development of novel therapeutic strategies.
O-GlcNAcylation: a novel therapeutic target for brain disorders
As discussed above, O-GlcNAcylation is implicated in various CNS disorders, including neurodevelopmental disorders, neurodegenerative diseases, psychiatric diseases, and CNS injuries. Understanding and harnessing the role of O-GlcNAcylation in the brain is crucial for identifying novel therapeutic targets for these diseases and injuries. Accumulating evidence has demonstrated the protective effects of O-GlcNAc modification in neurological diseases (see Table 2).
Increasing O-GlcNAcylation is protective against cognitive impairments and the pathogenic processes of neurodegenerative diseases. Reports indicate that enhancing neuronal OGT levels in the hippocampus can rescue age-related cognitive decline [34]. Tau protein is extensively O-GlcNAcylated, and this modification competes with phosphorylation, effectively reducing tauopathy [41, 178, 179]. Additionally, O-GlcNAcylation of APP has been shown to reduce β-Amyloid peptide production and amyloid plaques formation [180]. Furthermore, O-GlcNAcylation of α-syn prevents its aggregation, thereby protecting neurons from PD [136,137,138, 181]. These studies suggest that O-GlcNAcylation may protect neurons from pathogenic impairment, and enhancing this modification may inhibit the progression of certain diseases. Therefore, manipulating O-GlcNAcylation of proteins is a promising therapeutic approach for neurological pathologies [152, 182].
Extensive preclinical validation has underscored the significant potential of O-GlcNAcylation as a therapeutic target in neurodegenerative diseases, sparking great research interest among multiple companies seeking OGA inhibitors. This has led to the discovery of a range of chemical compounds suitable for further study in clinical trials. Pharmacological studies have demonstrated that OGA inhibitors can effectively elevate O-GlcNAcylation, decrease amyloid aggregates formation, and reduce neuronal death in animal models of AD and PD [178, 180, 183, 184]. Notably, TMG has been demonstrated to reduce Tau phosphorylation, thereby curbing Tau aggregation and neurodegeneration [185]. Additionally, it restores memory and cognitive function, enhancing spatial working memory and novel object recognition in the 5xFAD mouse model [186, 187]. Another significant study highlighted that ASN90 (also referred to as ASN-120290 or ASN-561), a novel OGA inhibitor, improved survival, respiratory function, and motor skills in the JNPL3/P301L mouse model of AD, attributed to an increase in O-GlcNAcylated Tau and a reduction in neurofibrillary tangles [183]. Furthermore, enhancing O-GlcNAcylation through GlcN injection ameliorated PD pathogenesis induced by 6-OHDA in mouse brains, with attenuated neuroinflammation and mitochondrial dysfunction and mitigated motor deficits [188].
Two compounds, TMG and GlcN, have been extensively investigated in the context of stroke. In mice models, it has been demonstrated that a moderate elevation of O-GlcNAcylation with TMG dramatically decreased infarct volumes, improved neurological deficits, and enhanced functional outcomes [50, 110, 189]. On the other hand, GlcN, a naturally synthesized amino monosaccharide produced within the body, effectively enhances UDP-GlcNAc production, subsequently leading to an elevation in global O-GlcNAcylation levels. Studies have shown that GlcN treatment elevates O-GlcNAcylation levels in the brain, exhibiting neuroprotective effects in both transient and permanent stroke models [50, 189]. These results indicate the positive effects of O-GlcNAcylation on stroke recovery.
In clinical study, several potent and selective OGA inhibitors have been developed as potential therapeutics [190]. Clinical studies have shown promising results for the treatment of AD and related tauopathies using MK-8719 [190,191,192] and ASN90 [190, 193]. Both drugs have been proven to be safe and well-tolerated with no noticeable adverse effects in phase I clinical trials. LY3372689, an OGA inhibitor developed by Eli Lily Co., is also being investigated as a promising treatment for tauopathies including AD [190, 194]. These data fully confirm the feasibility of OGA inhibition as an exciting and promising novel target for the treatment of neurodegenerative diseases, exhibiting potential therapeutic effects and broad application prospects.
In parallel with efforts to explore the potential therapeutic benefits of OGA inhibitors, researchers have also been investigating radiolabeled analogs to assess the target engagement and enzyme occupancy of potential therapeutic agents using PET. This will facilitate the therapeutic development of OGA inhibitors. One such PET radioligand, 18F-LSN3316612, has been reported to be selective and possess a high affinity for binding with OGA, making it excellent for imaging and quantifying OGA in animal models [195].
Although promising preclinical and clinical results have been reported, the clinical application of such chemical compounds remains fraught with limitations. Firstly, the physiological and pathological functions of O-GlcNAcylation are not fully understood under certain conditions, necessitating further exploration. Secondly, the specificity of chemical inhibitors must be carefully considered. Given that O-GlcNAcylation is a widespread intracellular PTM, inhibitors altering O-GlcNAcylation can impact thousands of proteins, many of which are not related to disease and may induce adverse effects. Recently, methods for specifically modifying O-GlcNAc on target proteins using gene editing [60], nanobodies [196] or nucleic acid aptamers [197] have been reported, suggesting increased potential for precision medicine in the future. Gene editing can introduce point mutations to specifically modify O-GlcNAcylation sites [60], Nanobodies can be fused with engineered OGT or splitOGA to install or remove O-GlcNAc on target proteins within living cells [196]. Aptamers, single-stranded DNA or RNA with unique three-dimensional conformations, tightly bind to target proteins, offering promise for targeted O-GlcNAc modification on specific proteins [197]. However, the application of these new techniques awaits more study. Thirdly, most chemical molecules targeting the O-GlcNAc pathway are not cell-permeable, limiting their delivery. Therefore, the development of highly specific and easily deliverable methods is essential and will require a significant investment of time and effort.
Conclusion and perspectives
O-GlcNAcylation emerges as a pivotal regulator of brain development and function, positioning it as an attractive therapeutic target for CNS disorders. This review has delineated the physiological and pathological roles of O-GlcNAcylation, elucidated the molecular mechanisms underpinning these functions, and underscored its protective potential in CNS diseases. The growing body of evidence supports O-GlcNAcylation’s critical involvement in neuronal survival, synaptic plasticity, and neuroprotection against various pathological conditions. However, several challenges remain before we can fully harness its therapeutic potential:
First, complexity of O-GlcNAcylation dynamics: O-GlcNAcylation plays a crucial regulatory role across various physiological and pathological processes. However, the simultaneous modification of numerous proteins under differing physiological and pathological conditions complicates the isolated analysis of O-GlcNAcylation on individual proteins. This requires careful, multifactorial approaches to avoid misinterpretation within the overall modification landscape.
Second, interplay with other PTMs: O-GlcNAcylation is known to interact with other PTMs, including phosphorylation and acetylation, through mutually influential or competitive mechanisms, contributing to the formation of a sophisticated “PTM network”. This network’s influence on critical neural activities such as neuronal function, memory formation, and synaptic plasticity is still under investigation, thus limiting the understanding of O-GlcNAcylation’s regulatory roles in neurological disorders. Clarifying these complex interactions is essential for accurately characterizing the functional implications of O-GlcNAcylation in the CNS.
Third, limitations of preclinical data and the need for clinical validation: Although these initial studies offer fundamental insights into O-GlcNAcylation’s biological functions, majority of the current data originates from cell and animal models, which may not fully reflect the human biological context. The limited availability of clinical data on O-GlcNAcylation presents a critical gap, rendering the translational relevance of preclinical findings uncertain. However, conducting clinical research on O-GlcNAcylation is fraught with technical and ethical challenges, highlighting an urgent need to bridge the gap between experimental models and clinical applications. This transition is crucial for achieving true translational impact in O-GlcNAcylation research and optimizing its potential therapeutic applications.
Fourth, the dysregulation of O-GlcNAcylation manifests diverse effects across various diseases, influenced by tissue and cell-type specificity, environmental and lifestyle factors, genetic and epigenetic variations, aging, and disease-specific signaling pathways. For example, aging and metabolic shifts can disrupt O-GlcNAcylation dynamics, playing a role in age-related conditions. In autoimmune and inflammatory diseases, O-GlcNAcylation regulates pathways like NF-κB, resulting in context-dependent effects. Therefore, a thorough exploration of these characteristics is crucial for unraveling the diverse roles of O-GlcNAcylation in disease mechanisms and for developing targeted therapeutic strategies.
To advance the field, future research should focus on:
First, developing highly specific tools to manipulate O-GlcNAcylation: Achieving high specificity in tools for selectively manipulating O-GlcNAcylation on individual proteins or within specific cell types is a critical objective. Current methodologies often lack the precision required to target distinct proteins or cellular environments, hindering efforts to isolate the specific role of O-GlcNAcylation in unique biological contexts. Developing tools capable of precise modulation at the level of individual proteins or specific cell populations, such as neurons or glial cells, would allow researchers to delineate the functional implications of O-GlcNAcylation with greater accuracy. Advanced techniques, including targeted gene editing, protein-specific inhibitors, and novel chemical probes, could significantly improve the precision of O-GlcNAcylation manipulation, thereby creating new opportunities for research and therapeutic applications.
Second, elucidating the complex interplay between O-GlcNAcylation and other PTMs in neuronal function and pathology: Investigating the intricate interactions between O-GlcNAcylation and other PTMs, such as phosphorylation, acetylation, and ubiquitination, is essential for understanding neuronal function and pathology. These modifications often interact in context-dependent and complex ways, contributing to a regulatory network that governs vital aspects of neuronal health, including synaptic plasticity, memory formation, and neuroprotection. Mapping PTM crosstalk in detail and identifying key regulatory pathways could reveal new insights into the molecular mechanisms underlying neurological diseases. This knowledge would establish a foundation for targeted strategies to modulate these interactions, potentially altering disease progression and improving neurological health.
Third, conducting well-designed clinical trials to validate the therapeutic potential: While preclinical studies indicate that modulating O-GlcNAcylation may hold promise for treating CNS disorders, carefully designed clinical trials are essential to validate these findings in humans. Such trials must rigorously evaluate the efficacy and safety of O-GlcNAcylation-targeting approaches in patients, with particular attention to CNS disorders where treatment options are limited. Clinical investigations could explore various therapeutic modalities, such as small molecules, gene therapies, or other interventions that selectively modulate O-GlcNAcylation levels in specific brain regions or neuronal populations.
Fourth, investigating potential off-target effects and long-term consequences: Given O-GlcNAcylation’s regulatory role in numerous cellular processes, unintended effects in non-targeted proteins or cells could disrupt essential signaling pathways, leading to adverse outcomes. Long-term consequences of O-GlcNAcylation modulation should be rigorously investigated, particularly regarding neuroplasticity, cellular resilience, and potential neurotoxicity. Studies using animal models and in vitro systems to explore the impact of sustained O-GlcNAcylation modulation will be critical to identifying and mitigating risks, ultimately leading to safer, more effective CNS-targeted therapies.
Fifth, exploring combination therapies targeting O-GlcNAcylation alongside other disease-modifying approaches: Combination therapies that target O-GlcNAcylation alongside other pathogenic mechanisms may enhance therapeutic efficacy in complex CNS disorders. Given that neurological diseases often involve multifactorial pathologies, targeting multiple pathways could yield synergistic effects, slowing disease progression and improving clinical outcomes. For example, therapies that modulate O-GlcNAcylation in conjunction with interventions targeting neuroinflammation, mitochondrial dysfunction, or protein aggregation could address several aspects of CNS pathology simultaneously. Rigorous preclinical and clinical studies are needed to identify promising combination strategies, thoroughly assess their safety and efficacy, and optimize dosing regimens to achieve maximal therapeutic benefit.
In conclusion, while O-GlcNAcylation holds promise as a therapeutic target for CNS disorders, advancing beyond animal and cell-based investigations toward rigorous clinical studies is imperative. This progression is crucial for translating the therapeutic potential of O-GlcNAcylation into tangible clinical benefits for patients with CNS disorders. As our understanding of this complex modification grows, so does the potential for developing novel, effective treatments for a wide range of neurological conditions.
Responses