Calboxyvinyl polymer adjuvant enhances respiratory iga responses through mucosal and systemic administration

Introduction
Vaccination is a highly effective strategy for controlling the spread of pathogens such as viruses and bacteria. Mucosal vaccine strategies are particularly favorable for respiratory viruses such as influenza and coronavirus1,2,3,4. Intranasal vaccines deliver antigens directly to the upper respiratory tract and nasal cavity, which are the sites where respiratory viruses attach and initiate infection3,4. The IgA antibodies present in these mucosal regions possess the ability to block viral infections5,6,7,8,9.
It is well-established that nasal vaccines can induce secretory IgA antibody production in the nasal cavity, which is rarely achieved through subcutaneous or intramuscular inoculation5,10. IgA antibodies have a higher neutralizing ability than IgG antibodies for certain antigens and exhibit high cross-reactivity to different virus strains, making them effective for protecting against virulent strains of viruses such as influenza and coronavirus10,11,12,13,14,15,16,17. Currently, seasonal influenza vaccines are mainly administered via the subcutaneous or intramuscular route and this strategy is effective at preventing serious infection from predicted circulating strains but is unable to prevent initial infection and has limited efficacy against unexpected strains and limited effectiveness in older individuals18,19,20. Therefore, developing a vaccine that can induce stronger and broader protection is desired. Nasal vaccines may have the potential to overcome these limitations. For example, the intranasal live attenuated influenza vaccine (Flumist®) is approved worldwide and has been shown to enhance mucosal and systemic immune responses, being particularly effective in children21. However, it is unsuitable for immunocompromised individuals22 and requires refrigerated transport23, making it less viable for use in developing countries. Thus, there is a need for stable alternative vaccines.
Adjuvants are compounds added to vaccines to enhance immunogenicity. Alum, the most widely used adjuvant in vaccines for humans24,25,26, is an insoluble particle and thus unsuitable for nasal delivery27. Nasal vaccines also require adjuvants that should be less invasive to the mucosa. Adjuvants targeting pattern recognition receptors (PRRs), such as poly I:C, R848, and CpG K3, have shown efficacy in nasal vaccines by primarily inducing T helper cells (Th)1 immunity, although Th2 immunity is also important for IgA production28,29.
While adjuvants can enhance vaccine immunogenicity, they also confer a risk of adverse effects such as swelling and fever30,31. Most adjuvants stimulate cytokine secretion from various cells, which although crucial for adjuvanticity, is involved in inflammation and can cause local and systemic side effects30,31. Therefore, developing highly safe adjuvants requires both immunological and safety data. Some PRR agonists show good adjuvanticity in mice but cause severe side effects in humans, as typified by poly I:C32. One approach to finding safe adjuvants is to repurpose approved drugs, pharmaceutical base materials, and food additives using appropriate screening systems.
We previously developed an in vivo screening system to predict and evaluate the adjuvanticity and toxicity of test compounds based on biomarker gene expression profiles in the lungs33,34,35. This system can predict and evaluate hematological toxicity, cytotoxicity, Th1/Th2 balance, and cytotoxic T lymphocyte (CTL) activation within 3 days35. Verification with existing adjuvants has shown the effectiveness of this system for safety and efficacy assessments.
In this study, we screened novel adjuvant candidates from 17 pharmaceutical base materials using our adjuvant screening system. We identified carboxy vinyl polymer (CVP) as a strong adjuvant. The biomarker gene expression profile of CVP suggests that it acts similarly to damage-associated molecular pattern (DAMP)-inducible adjuvants. The CVP-added influenza split vaccine (SV) induced approximately 20–1000 times higher antigen-specific IgA production than existing adjuvants such as alum and poly I:C with a single vaccination, successfully protecting mice from influenza virus infection. CVP also showed strong adjuvanticity with the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spike protein S1 antigen successfully protecting mice from SARS-CoV-2 infection. Additionally, the immunogenicity of the CVP-containing vaccine was evaluated using cynomolgus monkeys.
Results
Exploration of adjuvant candidates from pharmaceutical base materials
We previously constructed an assay system to evaluate adjuvanticity based on the expression profiling of biomarker genes in the lungs35. In this study, using this assay system, we explored substances with adjuvanticity for nasal vaccines from pharmaceutical-based materials. The pharmaceutical base materials included 17 types of nasal drop base materials, the compositions of which are shown in Table S1. These base materials were nasally administered to mice at a four-fold dilution in saline.
To conduct the screening, test compounds were intranasally administered to mice, and 16 h after administration, lungs were collected to analyze biomarker gene expression levels. Existing adjuvants, namely alum, AddaVax, poly I:C, R848, and CpG K3, were also used as controls. The results are displayed as a heatmap of biomarker gene expression levels (Fig. 1A). As shown in Fig. 1A, mice administered the toxicity reference influenza vaccine (RE), poly I:C, R848, or CpG K3 exhibited elevated expression of most biomarker genes in the lungs, consistent with their known adjuvanticity and high immunogenicity via type 1 interferon (IFN)-mediated immunity as previously reported35. Interestingly, base materials #1–8 showed marked elevation of Timp1, Cxcl11, and Cxcl9 (Fig. 1A and Supplementary Fig. 1A). According to a previous report35, a marked increase in Timp1 expression is indicative of mucosal adjuvanticity, as it is associated with inducing the antigen-specific IgA antibody production level in nasal vaccines, suggesting that base materials #1–8 confer adjuvanticity in nasal vaccines.

A Mice were intranasally administered various influenza vaccines, pharmaceutical base materials, or existing adjuvants. Lungs were harvested 16 h post-administration, and biomarker gene expression levels were measured by the QuantiGene Plex assay (QGP) method. The far left column indicates the administered samples, and the values in the cells represent the relative gene expression changes normalized to 1 for the saline (SA) control. Based on previous reports, the adjuvanticity and toxicity of each evaluated sample were predicted from gene expression changes according to the function specified by each biomarker gene. B Prediction of adjuvanticity based on biomarker gene analysis. The predicted values were calculated by biomarker gene expression levels in the lungs according to the method described in a previous study, which is detailed in the “Materials and Methods” section. Each column presents the mean value from two independent experiments.
To estimate the mode of action of their adjuvanticity, the biomarker gene expression profiles were analyzed. The results indicated that base materials #1–8 conferred adjuvanticity as evidenced by increasing IgA antibody production (Fig. 1B). Hierarchical clustering analysis was performed to predict the mode of action of the adjuvants based on the expression fluctuation patterns of the biomarker genes (Fig. S1B). The results indicated that the biomarker gene expression patterns of base materials #1–8 were similar to those of alum and AddaVax (Supplementary Fig. 1B). Since alum and AddaVax are known to induce slight cytotoxicity and are DAMP-inducible adjuvants36,37, it is speculated that base materials #1–8 are also DAMP-inducible adjuvants. Taken together, these results suggest that base materials #1–8 are potential adjuvant candidates.
Assessment of the adjuvanticity and physical characteristics of the pharmaceutical base materials
To assess the adjuvanticity of the test base materials, vaccine antigen-specific IgA antibody production in the base material-containing vaccines was analyzed. Mice were intranasally inoculated with SV, with or without each base material, and 21 days after inoculation, bronchoalveolar lavage fluid (BALF) was collected to assess antigen-specific IgA antibody production levels. The results showed that vaccine antigen-specific IgA antibody production levels were significantly and markedly increased when pharmaceutical base materials #1–8 were individually added to the SV compared with the SV alone (Fig. 2A). Furthermore, the antigen-specific IgA antibody production levels with base materials #1–8 combined with SV were higher than those with poly I:C, CpG K3, R848, AddaVax, or alum–adjuvanted SV or non-adjuvanted whole-particle inactivated influenza vaccine (WPV) (Fig. 2A). As described in the Materials and Methods section, the dosage of the adjuvant was determined by referencing reports in which it was used as an intranasal vaccine adjuvant in mouse experiments. These results indicate that base materials #1–8 have stronger adjuvanticity than some existing adjuvants in nasal vaccines. However, comparisons with toxin-derived adjuvants, such as cholera toxin B subunit (CTB) and heat labile toxin of enterotoxigenic Escherichia coli (LTB), have not been conducted, and it remains unclear whether CVP is superior to these mucosal adjuvants. Evaluating the potential of CVP as an adjuvant through comparisons with CTB and LTB represents an important future research objective.
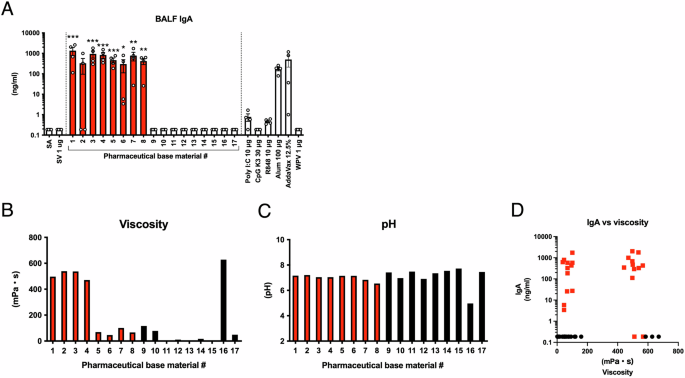
A Mice were intranasally administered saline (SA), split influenza vaccine (SV) alone, or SV with either pharmaceutical base materials (final 50% v/v) or existing adjuvants in a single dose. Three weeks post-administration, bronchoalveolar lavage fluid (BALF) was collected, and vaccine antigen-specific IgA antibody levels were measured by ELISA. The red column indicates pharmaceutical base materials that exhibited a marked increase in the mean IgA antibody titer. B Viscosity of pharmaceutical base materials diluted to 50% v/v in PBS. The red column indicates pharmaceutical base materials that exhibited a marked increase in the mean IgA antibody titer. C pH of pharmaceutical base materials diluted to 50% v/v in PBS. The red column indicates pharmaceutical base materials that exhibited a marked increase in the mean IgA antibody titer. D Correlation between viscosity and IgA antibody levels in BALF. The red dots represent pharmaceutical base materials that demonstrated a marked increase in the mean IgA antibody titer. Each dot represents data from an individual animal (n = 4) from two independent experiments. A Bar graphs indicate mean values and error bars represent the SEM of data from two independent experiments. B and C indicate the average value of two independent measurements. *p < 0.05, **p < 0.01, and ***p < 0.001 by Dunnett’s multiple comparison test following the Kruskal–Wallis test.
Pharmaceutical base materials serve to impart viscosity and pH stability in formulations. When the viscosity and pH of the 17 test pharmaceutical base materials were analyzed, no correlation was observed with IgA antibody-inducing ability (Fig. 2B–D). These results suggest that the adjuvanticities of base materials #1–8 are independent of their viscosity.
Pharmaceutical base material adjuvanticities are induced by CVP
As shown in Table S1, the component commonly contained in base materials #1–8, expected to have strong adjuvanticity, is CVP. CVP is included in some pharmaceutical base materials as a highly safe thickener, consisting of cross-linked polyacrylic acid. To demonstrate the adjuvanticity of CVP, mice were nasally inoculated with SV, with or without CVP or non-cross-linked CVP (Ncl-CVP), and the biomarker gene expression levels and immunogenicity were analyzed. Mice were immunized on days 0 and 21, and samples were collected on day 35. Biomarker gene expression analysis was performed using lung samples collected 16 h after a single vaccination. The results showed that Ncl-CVP did not induce Timp1, Cxcl9, or Cxcl11 expression levels as a marker for adjuvanticity (Fig. 3A) and showed little enhancement of antigen-specific IgA antibody production (Fig. 3B). By contrast, CVP administration increased Timp1, Cxcl9, and Cxcl11 expression levels (Fig. 3C) and antigen-specific IgA antibody production (Fig. 3D), indicating that CVP has adjuvanticity in nasal vaccines. Furthermore, IgA production levels were higher than those with existing adjuvants (Fig. 3E). We also evaluated high-viscosity CVP (HV-CVP). The HV-CVP was administered at lower concentrations than CVP based on its extremely high viscosity, which posed challenges for formulation preparation and intranasal administration in mice. Despite the lower concentration, vaccines containing HV-CVP increased IgA antibody production comparable to that induced by CVP (Fig. 3D). Because of the differences in concentrations, it was not possible to determine whether CVP or HV-CVP exhibits superior adjuvanticity. However, the results suggest that both CVP and HV-CVP have strong potential as vaccine adjuvants. In subsequent experiments, we focused exclusively on CVP because of the difficulties associated with the preparation and nasal administration of HV-CVP.
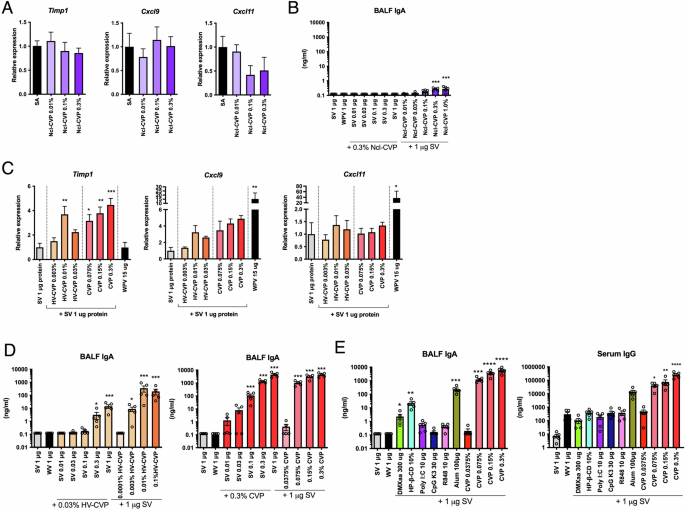
Mice were intranasally administered each CVP, and lungs were harvested 16 h later. Biomarker gene expression levels were measured by the QuantiGene Plex assay (QGP) method (A and C). Mice were intranasally administered saline (SA), split influenza vaccine (SV) alone, or SV with each CVP or existing adjuvants in a single dose. At 3 weeks post-administration, bronchoalveolar lavage fluid (BALF) and blood were collected, and vaccine antigen-specific IgA and IgG antibody levels were measured by ELISA (B, D, and E). Each dot represents an individual animal. Bar graphs indicate mean values and error bars represent the SEM (n = 5) of data from two independent experiments. *p < 0.05, **p < 0.01, and ***p < 0.001 by Dunnett’s multiple comparison test following the Kruskal–Wallis test (compared with the far left column).
Furthermore, we investigated whether CVP exhibits adjuvanticity in subcutaneously administered vaccines. The results showed that CVP significantly increased antigen-specific IgG antibody titers in the serum, even with subcutaneous vaccine administration (Supplementary Fig. 2). These findings indicate that CVP possesses strong adjuvanticity not only in mucosal vaccines but also in injectable vaccines.
CVP-adjuvanted influenza vaccine exhibits CTL activation in mice
To evaluate whether the adjuvanticity of CVP can induce vaccine antigen-specific CTLs, we conducted a series of experiments. Mice were vaccinated with the antigen alone or combined with various adjuvants, delivered via the intranasal or intramuscular route, with two vaccinations 3 weeks apart. Ten days after the booster, the spleen and lungs were harvested, and lymphocytes were isolated. These lymphocytes were then stimulated in vitro with the vaccine antigen, and the number of IFNγ-producing cells was assessed using an ELISPOT assay. In the intramuscularly vaccinated mice, we observed an increase in IFNγ-producing cells in those vaccinated with both poly I:C and CVP (Fig. 4A). However, in the intranasally vaccinated mice, a slight increase in IFNγ-producing cells was observed with poly I:C but not with CVP (Fig. 4A). To directly evaluate CTL activity, an in vivo killing assay was performed. Mice were vaccinated as described above, and 10 days post-boost, spleen cells pulsed with the vaccine antigen and labeled with a fluorescent marker were intravenously transferred into the immunized mice. Twenty-four hours post-transfer, the spleen and lungs were collected, and the fluorescently labeled target cells were counted to assess specific killing by CTLs. The results indicated that intramuscular vaccination with CVP exhibited the highest CTL activity, whereas intranasal vaccination with CVP showed CTL activity comparable to that with poly I:C (Fig. 4B). This discrepancy with the ELISPOT results suggests that CVP may induce CTL responses via an IFNγ-independent pathway when used in intranasal vaccines.
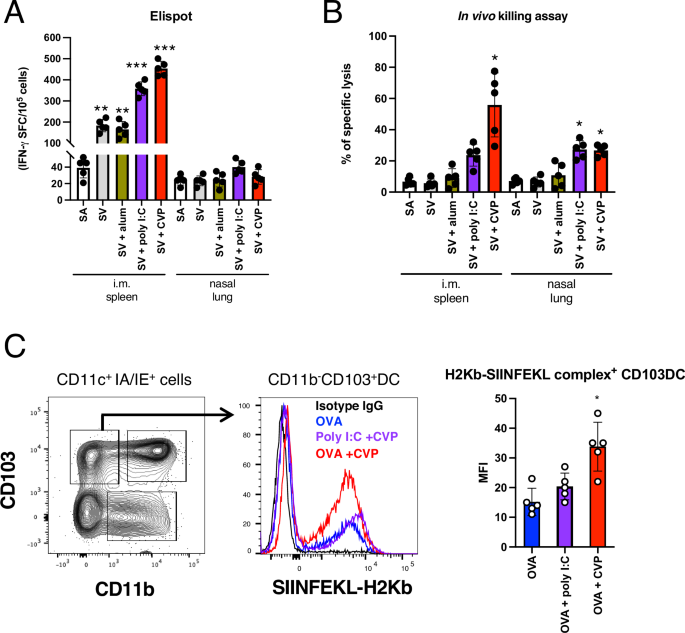
A Mice were intranasally or intramuscularly administered saline (SA), split influenza vaccine (SV) alone, or SV with each CVP or existing adjuvants twice, with a 4-week interval. Ten days after the boost, spleens or lungs were harvested, and lymphocytes were purified. Purified lymphocytes were incubated with SV antigen, and IFNγ-producing cells were measured by ELISPOT following overnight antigen stimulation. B Mice vaccinated with the same protocol above were intravenously injected with CFSE-labeled splenocytes pulsed with antigen from untreated mice. Twenty-four hours post-transfer, spleens or lungs were collected, and the frequency of CFSE-labeled transferred cells was measured by FACS to assess specific killing. C Lungs from mice immunized with OVA antigen using the same protocol were collected, and lymphocytes were purified. Purified lymphocytes were stained with antibodies against surface antigens and antibodies recognizing the OVA antigen peptide–H2Kb complex. The frequency of antigen-presenting cells was analyzed by FACS. Each dot represents an individual animal. Bar graphs indicate mean values and error bars represent the SEM (n = 5) of data from two independent experiments. *p < 0.05, **p < 0.01, and ***p < 0.001 by Dunnett’s multiple comparison test following the Kruskal–Wallis test (compared with the SA or OVA group).
Next, we assessed cross-presentation activity, which is important for CTL induction. Certain dendritic cells (DCs) are known to activate CD8+ T cells through major histocompatibility complex (MHC) class I presentation38,39. Using ovalbumin (OVA), which has well-defined MHC class I-presented peptide sequences in mice, as a model antigen, we employed antibodies recognizing the MHC class I–OVA peptide complex to analyze antigen-presenting cells. Our results showed that CD11b–CD103+ DCs exhibited increased MHC class I antigen presentation when CVP was added (Fig. 4C). This finding indicates that CVP enhances MHC class I antigen presentation, thereby potentially contributing to the induction of CTLs.
CVP as a safe adjuvant with low cytokine induction properties
To assess the safety of intranasally administered CVP, leukopenic toxicity and body weight gain were evaluated. Mice were intranasally administered various concentrations of CVP, existing adjuvants, or WPV, and 16 h after administration, body weight changes and white blood cell (WBC) counts were analyzed. The results showed that although WPV or R848 administration significantly reduced body weight compared with the saline (SA) only control group, neither HV-CVP nor CVP induced significant body weight reduction (Fig. 5A). It has been reported that type 1 IFN-inducible vaccines and adjuvants can cause leukopenic toxicity. Mice administered WPV or R848 showed significant reductions in WBC counts compared with the SA group (Fig. 5B). However, WBC counts were not significantly reduced by CVP administration compared with the SA group (Fig. 5B). These results indicate that intranasal CVP inoculation does not cause leukopenic toxicity or severe systemic toxicity in mice.
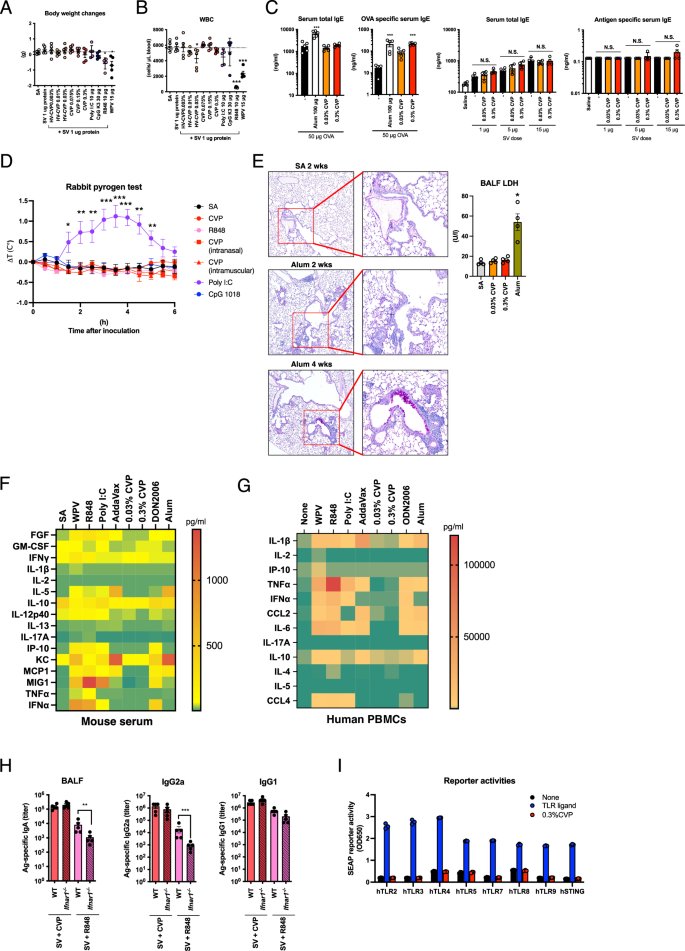
Mice were intraperitoneally injected with saline (SA), split influenza vaccine (SV) alone, or SV with either CVP or existing adjuvants. Body weight changes (A) and peripheral blood leukocyte counts (B) were analyzed 24 h post-injection. Mice were intranasally administered OVA antigen or SV with alum or CVP twice, with a 3-week interval, and serum antigen-specific or total IgE levels were measured 2 weeks post-boost (C). Rabbits were intravenously injected with CVP or various adjuvants, and rectal temperature changes were recorded (D). Mice received nasal or intramuscular injections of CVP alone. Mice were nasally administered alum or CVP at a dose of 50 µl/mouse, and bronchoalveolar lavage fluid (BALF) was collected 24 h later to measure lactate dehydrogenase (LDH) activity (E). Lungs were collected 4 weeks post-injection for PAS staining and histopathological analysis (E). Mice were intraperitoneally injected with SA, SV alone, or SV with either CVP or existing adjuvants. Blood was collected 24 h post-injection to measure serum cytokine levels (F). Human PBMCs were cultured with various vaccines, CVP, or adjuvant-containing media for 24 h, and cytokine levels in the supernatants were measured (G). Wild-type and Ifnar1˗/˗ mice were nasally administered SV with either CVP or R848, with a 3-week interval, and BALF and serum were collected 2 weeks post-boost to measure IgA and IgG antibody levels by ELISA (H). Human TLR or STING reporter cells were cultured with control ligands or 0.3% CVP for 24 h, and SEAP reporter activity was measured post-culture. Cytokine levels (F and G) are shown as heatmaps representing the mean values from three mice or from the PBMCs of three human donors. Each dot represents an individual animal (A–E and H: n = 5; E: n = 4; F, G, and I: n = 3). Bar graphs show the mean values and error bars represent the SEM of data from two independent experiments for A–H and from three independent experiments for I. A–E: *p < 0.05, **p < 0.01, and ***p < 0.001 by Dunnett’s multiple comparison test following the Kruskal–Wallis test (compared with the SA group or the left-most column). H: *p < 0.05 by the Mann–Whitney U-test. N.S. indicates not significant.
Some adjuvants, such as alum, are known to induce IgE antibody production, potentially leading to allergic reactions upon vaccination40,41. To assess whether CVP induces antigen-specific IgE antibody production, CVP was intranasally administered along with OVA, a model allergen antigen. The results showed that CVP enhanced antigen-specific IgE antibody production in serum (Fig. 5C). However, no antigen-specific IgE antibody was detected in serum when administered with SV at an effective antigen dose. Furthermore, the total serum IgE levels were not markedly elevated by CVP administration compared with the antigen alone, with the exception of the 1 µg SV immunization group (Fig. 5C). These results suggest that while CVP can induce antigen-specific IgE antibody production when OVA is used as an antigen, it does not do so at the dosing volume of influenza vaccine antigens sufficient for eliciting immunogenicity and protection. In addition, CVP did not significantly increase total serum IgE levels following OVA or SV antigen immunization.
One of the adverse reactions associated with adjuvants is fever30,31. Rabbits are particularly useful for assessing febrile responses because they readily exhibit fever when exposed to endotoxins or poly I:C42,43. Various adjuvants or CVP were intravenously administered to rabbits, with CVP also being administered intranasally and intramuscularly. The results indicated that CVP did not induce the fever response observed with poly I:C (Fig. 5D). Next, we pathologically evaluated the impact of CVP on the alveolar mucosa. Although intranasal vaccines typically do not directly reach antigen in the lungs, it is crucial to assess the toxicity to the lungs and bronchi for mucosal vaccine safety. We administered 25 µl of CVP or alum intranasally into each nostril of mice and evaluated lung tissue damage. Four weeks post-administration, alum caused the formation of lymphoid follicles and goblet cell hyperplasia, whereas these findings were not observed with CVP (Fig. 5E). In addition, lactate dehydrogenase (LDH) activity in BALF collected at 24 h post-vaccination was not elevated in CVP vaccinated mice (Fig. 5E). This suggests that CVP has a lower potential for inducing mucosal injury and chronic inflammation in the lungs.
Cytokine storms are often triggered by substances with high adjuvanticity31,44. To determine whether CVP could induce a cytokine storm, we administered CVP and various adjuvants intraperitoneally to mice and measured serum cytokine levels after 24 h. Inflammatory cytokine, IFN, and chemokine levels were elevated by some adjuvants; however, CVP did not induce marked production of any cytokines or chemokines (Fig. 5F, Supplementary data Table 2). Similarly, in human peripheral blood mononuclear cells (PBMC), CVP stimulation did not result in significant cytokine or chemokine secretion (Fig. 5G, Supplementary data Table 3). These findings suggest that the adjuvanticity of CVP is likely independent of cytokine induction. To further investigate the involvement of type 1 IFN signals in CVP adjuvanticity, we used type 1 IFN receptor knockout mice. Neither IgA nor IgG antibody responses to CVP-adjuvanted vaccine were attenuated in these knockout mice (Fig. 5H). By contrast, R848, known for its type 1 IFN signal-dependent adjuvanticity, showed reduced adjuvanticity in the knockout mice. These results demonstrate that CVP has a low potential to induce cytokine production and does not rely on type 1 IFN signals for its adjuvanticity. Subsequently, we investigated whether CVP directly activates PRR signaling using reporter cells expressing human PRR. The results showed that CVP did not activate stimulator of interferon genes (STING) or any Toll-like receptor (TLR) signaling pathways (Fig. 5I). These findings suggest that the adjuvanticity of CVP is elicited through a pathway independent of direct stimulation of TLRs or STING.
Single vaccination with CVP-adjuvanted influenza vaccine confers protection against lethal influenza virus infection
To assess the adjuvanticity of CVP, the protective effects of CVP-adjuvanted influenza vaccine were analyzed using a lethal influenza virus infection model. Mice were intranasally inoculated with SV with or without adjuvant, and WPV alone was used as a control. As shown in Fig. 2A, CVP induced potent IgA production following a single immunization. Therefore, a single immunization regimen was employed for the protection study. Non-treated mice served as negative controls. At 21 days after single vaccination, mice were infected with A/California/7/2009 [A(H1N1)pdm09] influenza virus at a dose of 5 × LD50 (50% of the lethal dose) (Fig. 6A). Body weight changes were monitored for 14 days post-infection. Nasal wash and BALF were collected at 3 days post-infection to assess virus titers. The results showed that mice inoculated with 0.3% CVP-adjuvanted SV did not exhibit body weight loss and significantly gained weight compared with the other groups (Fig. 6B). Although mice inoculated with 0.03% CVP-adjuvanted SV showed some weight loss, their recovery was faster than that of the SV, WPV, and non-treated groups (Fig. 6B). Mice inoculated with WPV or SV showed slower recovery compared with those inoculated with CVP-adjuvanted SV. The influenza virus titers in BALF were lower in the 0.3% CVP-adjuvanted group than in the WPV, SV, SV with R848, or poly I:C-adjuvanted groups (Fig. 6C). SV with alum showed similar influenza virus titers to the 0.3% CVP-adjuvanted SV group. By contrast, influenza virus titers in the nasal wash were reduced only in the CVP-adjuvanted SV group (Fig. 6D). These results indicate that a single vaccination of CVP-adjuvanted SV provides higher protective effects than some existing adjuvants combined with SV.
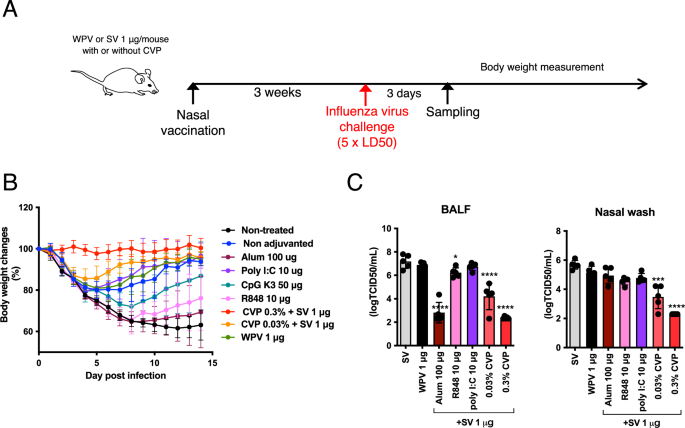
Mice were given a single nasal administration of split influenza vaccine (SV) alone, SV with various adjuvants or CVP, or whole-particle inactivated influenza vaccine (WPV), followed by nasal infection with A/California/07/09 H1N1 influenza virus at 5 × LD50 3 weeks later (A). Some mice were euthanized 3 days post-infection to collect BALF and nasal lavage fluids for viral titer measurement (B). Body weight changes were recorded for 14 days post-infection (C). Each dot represents an individual animal. Bar graphs represent mean values and error bars indicate the SEM (B: n = 10 and C: n = 5) of data from two independent experiments. *p < 0.05, ***p < 0.001, and ****p < 0.0001 by Dunnett’s multiple comparison test following the Kruskal–Wallis test (compared with the SA group).
Adjuvanticity of CVP on recombinant SARS-CoV-2 S1 protein vaccine
Nasal vaccines are highly effective in preventing viral infections in the nasal cavity and nasopharynx. SARS-CoV-2 infects these regions, facilitating human-to-human transmission and its pandemic potential. Thus, we investigated the utility of CVP in a nasal SARS-CoV-2 vaccine using the recombinant Wuhan strain SARS-CoV-2 S1 protein as the antigen. We assessed serum IgG and BALF/nasal wash IgA titers up to 150 days after two intranasal doses. The results showed that the CVP-adjuvanted nasal vaccine achieved the highest IgA titers, with sustained serum IgG and BALF/nasal wash IgA titers even 150 days post-boost (Fig. 7A). Interestingly, intramuscular administration of the CVP-containing vaccine also elevated BALF and nasal wash IgA titers, though these decreased significantly by 150 days post-boost (Fig. 7B). These findings suggest that the CVP-adjuvanted vaccine is highly effective against SARS-CoV-2, and CVP can also enhance local mucosal immunity when administered intramuscularly.
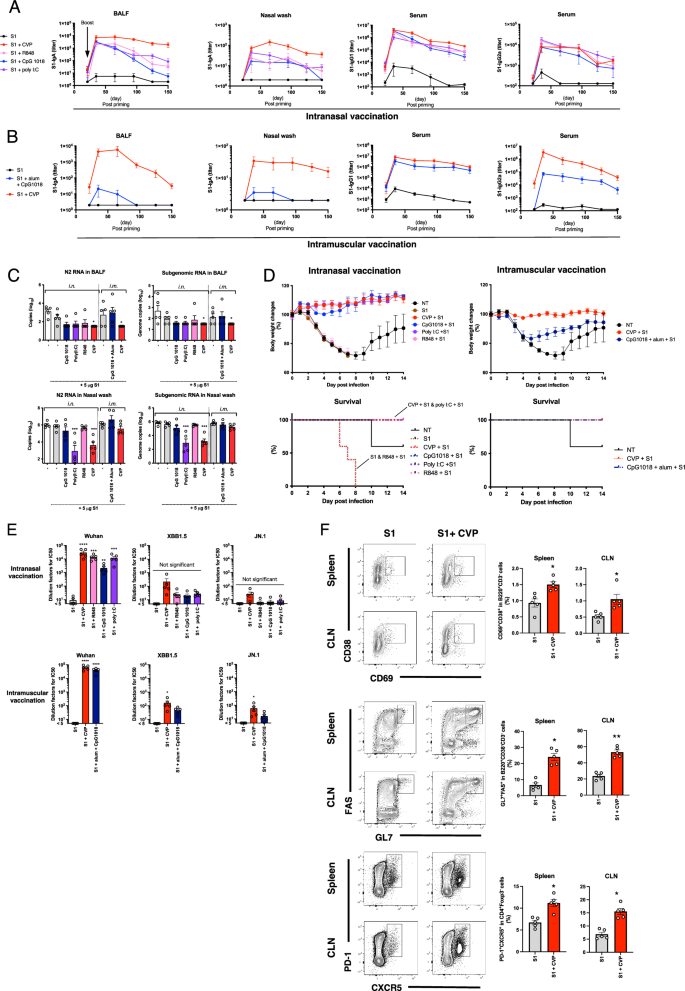
Mice received two nasal or intramuscular injections of SARS-CoV-2 S1 alone or S1 with various adjuvants or CVP, with a 4-week interval, followed by euthanasia at various time points post-boost to collect nasal and bronchoalveolar lavage fluids for measuring IgA and IgG antibody levels by ELISA (A and B). Some mice were nasally infected with mouse-adapted SARS-CoV-2 QHmusX virus (from a European lineage) 2 weeks post-boost, and some were euthanized 2 days post-infection to collect lavage fluids for RT–PCR-based viral RNA copy number measurement (C). Body weight changes and survival were monitored for 14 days post-infection (D). Serum collected at 14 days post-boost in (A) was used to measure neutralizing activity against SARS-CoV-2 Wuhan, XBB1.5, and JN.1 variants using a pseudovirus neutralizing assay (E). Lymphocytes isolated from the lungs of mice at 3 days post-boost immunized under the same conditions as in (A) were analyzed by FACS to determine the frequency of germinal center B cells and Tfh cells (F). Each dot represents an individual animal. Bar graphs show the mean values and error bars represent the SEM (A–C, E, and F: n = 5 and C: n = 10) of data from two independent experiments. *p < 0.05, **P < 0.01, ***p < 0.001, and ****p < 0.0001 by Dunnett’s multiple comparison test following the Kruskal–Wallis test (A, B, E, and F: compared with the S1 group and C and D: compared with the non-treated (NT) group). D The log-rank (Mantel–Cox) test was used to compare survival curves (**p < 0.01 compared with the NT group).
Next, we evaluated the protective efficacy of the CVP-adjuvanted vaccine against SARS-CoV-2 using a mouse-adapted SARS-CoV-2 QHmusX strain derived from the D614G (B.1) variant as previously reported45. Mice received two doses of various vaccines at 3-week intervals, followed by nasal infection with QHmusX virus 2 weeks post-boost. We monitored body weight and euthanized some of the mice at 48 h post-infection to collect BALF and nasal washes. The CVP-adjuvanted nasal vaccine significantly prevented SARS-CoV-2-induced weight loss and notably reduced subgenomic RNA and N2 RNA levels in BALF and nasal washes (Fig. 7C, D). These results demonstrate that the CVP-adjuvanted vaccine provides strong protection against infection and exhibits superior adjuvanticity compared with existing adjuvants.
Given the antigenic variation of SARS-CoV-2 resulting from amino acid mutations, we assessed the virus-neutralizing activity in sera from vaccinated mice using vesicular stomatitis virus (VSV) pseudotyped with SARS-CoV-2 spike protein. The results revealed that the sera from mice vaccinated with the CVP-containing vaccine exhibited the highest neutralizing titers against the Wuhan strain, while also maintaining weak neutralizing titers against the XBB.1.5 and JN.1 variants but the titers were not significantly increased following intranasal vaccination compared with S1 antigen alone (Fig. 7E). These findings suggest that the CVP-containing SARS-CoV-2 vaccine confers a weak cross-protective effect against various viral variants.
The production of high-quality antigen-specific antibodies is critically dependent on the maturation of B cells within the germinal center (GC) and the support provided by T follicular helper (Tfh) cells46,47. To investigate this, we performed fluorescence-activated cell sorting (FACS) analysis on GC B cells and Tfh cells in the lymph nodes and spleen of mice immunized with a CVP-containing intranasal vaccine. The results revealed that, compared to immunization with the S1 antigen alone, the CVP-containing intranasal vaccine induced a marked increase in the populations of activated non-GC B cells, GC B cells, and Tfh cells (Fig. 7F). These findings suggest that the CVP-containing intranasal vaccine enhances the maturation of B cells, thereby promoting the production of antigen-specific antibodies.
Robust immunogenicity of CVP-containing SARS-CoV-2 vaccine in cynomolgus monkeys
An immunogenicity test was performed on cynomolgus monkeys to evaluate the effectiveness of the CVP-containing vaccine in a model closely related to humans. The cynomolgus monkeys received the CVP-containing SARS-CoV-2 (Wuhan strain) spike trimer vaccine either intranasally or intramuscularly, with periodic collection of blood and nasal lavage samples (Fig. 8A). Analysis of antigen-specific IgA antibody titers in nasal lavage fluid showed a significant increase post-boost, with high levels maintained for 77 days (Fig. 8B). Remarkably, IgA antibody titers were also detected in nasal lavage fluid from monkeys vaccinated intramuscularly (Fig. 8B). Elevated serum IgG antibody titers were observed in the CVP-containing vaccine group (Fig. 8B). Monkeys vaccinated without CVP showed significantly lower IgA and IgG antibody levels. Neutralization tests using the Wuhan strain revealed high virus-neutralizing activity on days 35 and 75 in all CVP-containing vaccine groups (Fig. 8C). These results indicate that the CVP-containing vaccine has high immunogenicity in cynomolgus monkeys. Additionally, the increase in nasal IgA titers following intramuscular vaccination suggests that CVP-containing vaccines can activate mucosal immunity via intramuscular administration.
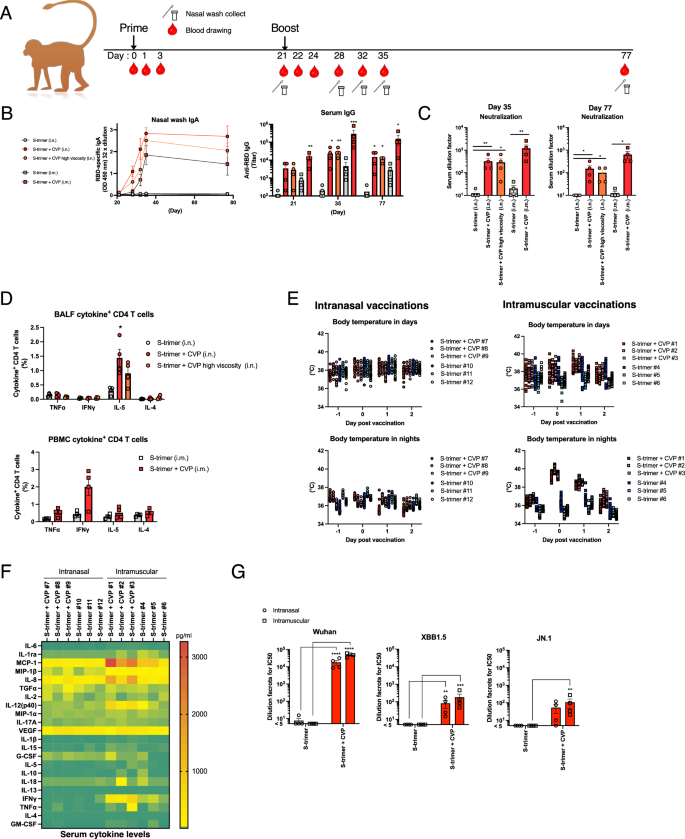
Cynomolgus monkeys received two nasal or intramuscular injections of SARS-CoV-2 S-trimer alone or S-trimer with CVP, with a 3-week interval (A). Serum and nasal lavage fluids were collected at various time points (A). IgA and IgG antibody titers were measured using collected nasal lavage fluids and serum (B), and serum neutralization titers against the SARS-CoV-2 Wuhan strain were determined (C). On day 35, lymphocytes and PBMCs were isolated from bronchoalveolar lavage fluid (BALF) and blood, stimulated overnight with S-trimer, and analyzed for intracellular cytokine levels by FACS (D). Body temperature changes post-vaccination were analyzed using temperature loggers implanted intraperitoneally in cynomolgus monkeys (E). The results are shown for 12-h day and night-time periods, respectively. Temperature data for each individual are represented by different colors. Temperature was recorded every 30 min, with 24 data points each for day and night-time. Data for two individuals in (E) are missing because of logger malfunction, therefore each group comprises three monkeys. Serum cytokine levels were measured on day 1 (F). Serum from day 35 was used to measure neutralizing activity against SARS-CoV-2 Wuhan, XBB1.5, and JN.1 variants using a pseudovirus neutralizing assay (G). Each dot represents an individual animal. Bar graphs show mean values and error bars represent the SEM (A–E and G: n = 4 and F: n = 3) of data from two independent experiments. *p < 0.05, **p < 0.01, and ***p < 0.001 by Dunnett’s multiple comparison test following the Kruskal–Wallis test (compared with the S-trimer group). C: **p < 0.01, ***p < 0.001, and ****p < 0.0001 by the Mann–Whitney U-test.
Subsequent analysis of cytokine production by CD4+ T cells on day 35 revealed an increase in interleukin (IL)-5-producing cells in BALF from intranasally vaccinated monkeys and an increase in IFNγ-producing cells in blood from intramuscularly vaccinated monkeys (Fig. 8D). Cytokine analysis in plasma showed no significant induction of cytokines following intranasal vaccination with the CVP-containing vaccine (Fig. 8E). However, increases in some Th1-related cytokines and some chemokines were observed following intramuscular vaccination (Fig. 8E). Body temperature monitoring via intraperitoneal loggers revealed no fever response in intranasally vaccinated monkeys, regardless of CVP presence, while a transient fever was observed in intramuscularly vaccinated monkeys with CVP (Fig. 8F, Supplementary data Table 4). These findings suggest that CVP-containing intranasal vaccines are unlikely to cause a cytokine storm or fever, while CVP-containing intramuscular vaccines may induce mild fever.
We assessed virus-neutralizing activity in sera from vaccinated mice using VSV pseudotyped with SARS-CoV-2 spike protein. The results revealed that the sera from cynomolgus monkey vaccinated with the CVP-containing vaccine exhibited the highest neutralizing titers against the Wuhan strain, while also maintaining weak neutralizing titers against the XBB.1.5 and JN.1 variants (Fig. 8G). These findings suggest that the CVP-containing SARS-CoV-2 vaccine confers a relatively higher cross-protective effect against various viral variants compared with some existing adjuvants.
Discussion
The primary targets of infection by influenza viruses and SARS-CoV-2 are the epithelial cells of the upper respiratory tract2,3,4,5,6. Therefore, inducing virus-specific nasal IgA antibodies on the nasal mucosal epithelium is beneficial. Nasal mucosal IgA antibody production has been shown to effectively suppress infection transmission, making it crucial in mitigating pandemics6,7,8. Nasal vaccination can induce IgA antibody production in the nasal mucosa and bronchi, which cannot be achieved through subcutaneous or intramuscular vaccination10. Many approved influenza and SARS-CoV-2 vaccines are administered via subcutaneous or intramuscular routes. While these vaccines can suppress the severity of infection symptoms, they cannot prevent primary infections in the nasal mucosa, bronchi, and lungs.
Numerous studies have reported on intranasal vaccines for influenza viruses7,8,9,10,11,12,13,14, demonstrating their ability to induce IgA antibody production and strongly inhibit influenza infection in the nasal mucosa and lungs. However, intranasal vaccination solely with antigen is insufficient to elicit appropriate immune responses in the upper respiratory tract11. Therefore, vaccine adjuvants are necessary to induce vaccine-specific nasal IgA responses. Adjuvants such as cholera toxin (CT) and heat-labile toxin (LT) from Escherichia coli have been used to boost the immunogenicity of nasal vaccines10,11,12. However, while CT and LT are effective adjuvants for enhancing mucosal immune responses, including secretory IgA responses, they have been associated with severe adverse effects in humans, such as Bell’s palsy48. Consequently, there is a need for adjuvants that are as effective as CT and LT but safer for human use in intranasal vaccines.
Several studies have demonstrated the utility of PRRs as vaccine adjuvants in mouse models24,25,26. TLR(s) agonists and STING agonists have shown enhancement of antibody production in mouse models30,32. However, some of these PRR agonists have raised safety concerns because of their association with the secretion of inflammatory cytokines32,33. Adjuvants targeting TLR7/8, such as R848, have been reported to induce cold-like symptoms in humans32,33. While these adjuvants demonstrate sufficient immunogenicity in mice, increasing the dosage raises the risk of adverse reactions in humans, making it challenging to achieve sufficient adjuvanticity. Thus, there is a need to develop novel adjuvants with both high efficacy and safety. Metal particle adjuvants, such as alum, are not suitable for intranasal vaccines as they can induce chronic inflammation in mucosal tissues and stimulate persistent IgE production27.
In this study, using the influenza virus SV, we explored the adjuvanticity of pharmaceutical base materials containing approved drugs and pharmaceutical excipients. Interestingly, we observed robust induction of vaccine antigen-specific IgA antibody production with pharmaceutical base materials containing the excipient component CVP, commonly found in nasal sprays (Fig. 3E). Adding CVP alone to nasal influenza vaccine-induced dose-dependent production of vaccine antigen-specific IgA antibodies, revealing the strong adjuvanticity of CVP (Fig. 3E). However, the basic structure of CVP, polyacrylic acid alone, resulted in lower levels of antibody production (Fig. 3B). This suggests that the cross-linked structure of polyacrylic acid is crucial for the adjuvanticity of CVP. The viscosity of vaccine formulations retains antigens in mucosal tissues that would contribute toward enhancing antigen uptake. While CVP-adjuvanted vaccine exhibits viscosity (Fig. 2B), among polymers with viscosity equal to or greater than that of CVP, none of the pharmaceutical base materials showed adjuvanticity (Fig. 2A, B, and D), indicating that viscosity alone cannot elicit the adjuvanticity of CVP. Moreover, intriguingly, CVP demonstrated strong adjuvanticity even following a single intranasal vaccination, resulting in significantly higher induction of IgA antibody production compared with alum- or PRR agonist-adjuvanted vaccines (Fig. 3E). However, no potent induction of antigen-specific IgE antibody production was observed with the influenza vaccine (Fig. 5C), suggesting that CVP is effective as a mucosal adjuvant for influenza vaccines. Unlike OVA, the lack of antigen-specific IgE production with SV (Fig. 5C) is likely attributable to the lower antigen dose in SV compared with OVA and the inherent ability of OVA to more readily induce antigen-specific IgE antibodies. Furthermore, total IgE levels did not increase with CVP administration in either the OVA or SV groups (Fig. 5C).
CVP has been used as a safe thickening agent in nasal drops49. We found that pharmaceutical base materials possessing the higher crosslinking density of CVP exhibited higher adjuvanticity. The effects of factors such as CVP concentration, molecular weight, particle size, and shear processing time on adjuvanticity require further investigation. The discovery of the adjuvant potential of CVP, a safe excipient, is a significant advancement.
Hierarchical clustering analysis of biomarker gene expression profiles has revealed similarities in the mode of action between CVP and existing adjuvants. Our results indicate that CVP acts as a DAMP induction-type adjuvant, unlike PRR agonists (Fig. S1B). This is supported by the observed increase in serum cytokines induced by PRR agonists, but not CVP administration (Fig. 5F). Furthermore, the leukopenic toxicity observed with type I IFN-inducing adjuvants, such as poly I:C or R848, was not induced by CVP administration (Fig. 5B). Additionally, the adjuvanticity of CVP was not diminished in type I IFN receptor knockout mice (Fig. 5H). These findings suggest that CVP adjuvanticity is less dependent on IFN and that CVP has a weaker propensity to induce a cytokine storm, indicating a possible reduction in the frequency of adverse reactions in humans.
In the case of intranasal vaccines, adjuvants may also reach the bronchi and alveoli. Metal particles, such as alum, have been reported to induce chronic inflammation and pneumonia in the lungs27. To verify this, mice were intranasally administered 25 µl of the sample per nostril, and lung damage and inflammation were subsequently assessed. This dosage ensures that the sample reaches the bronchi and lungs. Mice administered alum intranasally showed the presence of lymphoid follicles even 4 weeks post-administration, along with increased proliferation of Goblet cells in bronchial epithelial cells, indicating persistent inflammation (Fig. 5E). However, no such pathological changes were observed in mice administered CVP (Fig. 5E). Thus, CVP is anticipated to be safe even if exposure to the lungs occurs accidentally. As CVP is already used as an excipient in nasal sprays, its safety in humans is presumed.
This study conducted analyses using not only the influenza vaccine but also the SARS-CoV-2 S1 antigen vaccine. CVP demonstrated strong induction of IgA antibody production against S1 antigen (Fig. 7A). Furthermore, in both upper and lower respiratory tract infection mouse models, CVP significantly suppressed weight loss due to infection and reduced virus levels in the lungs and nasal cavity (Fig. 7C). Remarkably, even when administered intramuscularly, the CVP-containing vaccine induced robust IgA antibody production in the BALF temporarily (Fig. 7B). These immunogenic responses were also observed in cynomolgus monkey studies (Fig. 8B, C). To assess the protective efficacy against SARS-CoV-2 variants, virus neutralization tests were performed by a pseudovirus neutralization assay using a VSV pseudovirus with a SARS-CoV-2 spike protein. The findings revealed that neutralizing titers against XBB.1.5 and JN.1 were retained (Fig. 7E). These results suggest that the CVP-containing vaccine offers broad cross-protective effects against various viral variants. The cross-protective capacity and mucosal IgA induction potential of the CVP-containing vaccine indicate its effectiveness in protecting against variant strains with antigenic changes, such as those observed with SARS-CoV-2 and influenza viruses, and in mitigating pandemic spread through transmission control.
The CVP adjuvant increased the populations of GC B cells and Tfh cells, which are considered critical for inducing antibody production (Fig. 7F). Interestingly, within the presumed Tfh cell population, a marked increase in the PD-1int subset was evident (Fig. 7F). Adoptive transfer studies that attempted to subset Tfh populations have found that a population similar to pre-Tfh cells that express intermediate levels of Th1 and Tfh markers (CXCR5int, PD-1int, Ly6clo, T-betint) possesses the most central memory potential50,51,52. Based on these findings, the PD-1int population increased by CVP is presumed to be similar to pre-Tfh cells. Detailed functional analyses of the cell population found in the present study are required in the future.
The in vivo killing assay results demonstrated that CVP has the ability to induce vaccine-specific CTLs (Fig. 4B). However, the capacity of CVP to induce antigen-sensitization-dependent IFNγ production varied depending on the route of administration (Fig. 4A), leaving the underlying mechanisms of CTL induction unclear. Further immunological analyses, considering the route of administration, are required in future research. Additionally, cross-presentation by DCs was suggested as a potential mechanism for CTL induction by CVP (Fig. 4C). Nevertheless, the involvement of CD4+ T cells and CD8+ T cells remains unknown, highlighting the need for detailed immunological studies in the future.
The in vivo killing assay results demonstrated that CVP has the ability to induce vaccine-specific cytotoxic T lymphocytes (CTLs) (Fig. 4B). However, the capacity of CVP to induce antigen-sensitization-dependent IFNγ production varied depending on the route of administration, leaving the underlying mechanisms of CTL induction largely unclear. Further immunological analyses, considering the route of administration, are required. Additionally, cross-presentation by dendritic cells (DCs) was suggested as a potential mechanism for CTL induction. Nevertheless, the involvement of CD4+T cells and CD8+T cells remains unknown, highlighting the need for detailed immunological studies in future research.
These findings imply that CVP activates strong mucosal immunity, although the mechanism remains unclear. These effects surpassed those of PRR agonist adjuvants, highlighting the superior adjuvant properties of CVP compared with many existing adjuvants. Given its use as a pharmaceutical base material, CVP holds promise both in terms of safety and efficacy as an adjuvant. Furthermore, from a drug repositioning perspective, vaccines utilizing CVP as an adjuvant might initiate clinical trials more expeditiously than those using novel compounds.
This study found that the adjuvanticity of polyacrylic acid was relatively low and that its crosslinking structure was crucial for the adjuvanticity of CVP (Fig. 3B, 3E). This adjuvanticity was identified through screening using biomarker gene-based screening. The screening utilized Timp1 expression levels as an indicator of IgA antibody production in nasal vaccines, showing a significant increase in Timp1 expression in CVP-inoculated mice (Fig. 1A and Supplementary Fig. 1). The Timp1 gene has been reported to be useful as a marker for IgA production induction of DAMP-induced adjuvants, as its expression is highly sensitive to tissue damage35. Indeed, alum and AddaVax have been shown to increase Timp1 expression (Fig. 1A and Supplementary Fig. 1), leading to significant IgA antibody production induction upon nasal administration. However, because of their strong cellular toxicity and potential for chronic inflammation in the lungs, alum and AddaVax have not been considered safe for nasal vaccines. By contrast, CVP did not induce chronic inflammation or strong cellular toxicity in the lungs even if it reaches the lungs (Fig. 5E), demonstrating excellent properties as a DAMP-induced adjuvant. Detailed elucidation of the adjuvant mechanism of CVP will be the subject of a future study.
One limitation of this study is the use of a 30 μl (15 μl for each nostril) vaccine volume for intranasal vaccination in mice, a volume capable of delivering the vaccine adjuvant to the lungs and bronchi. Additionally, the analyses of antibody production and immune cells in this study primarily used lung cells and bronchoalveolar lavage fluid. Consequently, the results strongly reflect the effects observed in the lungs, which may differ from the responses elicited by intranasal vaccines in humans. In humans, intranasal vaccines do not reach the lungs. In the cynomolgus monkey experiments conducted in this study, a volume that does not reach the lungs was used for intranasal administration, demonstrating that CVP also exhibits adjuvanticity in nasal cavity-restricted immunity (Fig. 8B). Future studies should evaluate the CVP combined vaccine efficacy and adjuvant mechanisms using intranasal doses of less than 5 μl in mice to better align with the conditions relevant for human intranasal vaccine development.
CVP did not induce fever in rabbits; however, intramuscular administration in cynomolgus monkeys resulted in fever induction (Figs. 5D and 8E). Fever is generally thought to be triggered by inflammatory cytokines. In the rabbit experiments, the vaccine formulation did not include antigens, likely leading to a lower level of innate immune cell activation compared with the cynomolgus monkey experiments, thereby preventing the induction of a febrile response. These findings suggest that while CVP itself does not possess fever-inducing activity, the combination of CVP with an antigen administered intramuscularly is likely to induce fever.
This study successfully identified CVP among pharmaceutical base materials with adjuvanticity equal to or greater than existing adjuvants. CVP has been used as a base material in nasal sprays and demonstrates high safety on mucosal surfaces in humans. Furthermore, as a nasal vaccine adjuvant, it exhibits superior induction of IgA antibody production and CTL activity, defending against influenza and SARS-CoV-2 infections. Additionally, the adjuvanticity shows low dependency on IFN and cytokines, suggesting a unique mechanism of action. With a history of human use, CVP holds promise for early practical application as a nasal vaccine adjuvant.
Materials and Methods
Antigens and adjuvants
The influenza A virus (A/California/7/2009; H1N1) SV and whole-particle vaccine were generously provided by the Kitasato Institute (Tokyo, Japan). WPV comprising three different types of inactivated whole-virion components: A/Newcaledonia/20/99 (H1N1), A/Hiroshima/52/2005 (H3N2), and B/Malaysia/2506/2004) used for leukopenic toxicity tests were obtained from KM Biologics Co., Ltd (Kumamoto, Japan). RE, issued by NIID (Japan), is used as a toxicity reference in the leucopenic toxicity test in Japan33. RE is a lyophilized whole-virion preparation of an inactivated influenza virus, consisting of three different types of inactivated whole-virion: A/Newcaledonia/20/99 (H1N1), A/Hiroshima/52/2005 (H3N2), and B/Malaysia/2506/2004. To make 1.0 U/0.5 ml of RE solution, freeze-dried RE was reconstituted in physiological saline (SA). SARS-CoV-2 (Wuhan strain) spike protein S1 with Fc-tag antigen and SARS-CoV-2 (Wuhan strain) spike trimer protein were obtained from ACROBio systems (Newark, DE, USA). The following commercially available adjuvants were used: aluminum hydroxide gel (Alhydrogel, alum), squalene-based oil-in-water adjuvant AddaVax, murine STING ligand DMXaa, Toll-like receptor 1/2 (TLR1/2) agonist Pam3CSK4, TLR3 agonist poly I:C, CpG ODN2006, and TLR7/8 agonist R848 obtained from InvivoGen (San Diego, CA, USA). The TLR9 agonist CpG K3 (B–type CpG ODN) was generously provided by Prof. Ken J Ishii (The Institute of Medical Science, The University of Tokyo, Tokyo, Japan). Low endotoxin OVA (<1.0 endotoxin unit/mg) was purchased from FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan). Prenylamine, trimipramine maleate salt, and povidone were from Sigma-Aldrich (St. Louis, MO, USA). Etoposide was from FUJIFILM Wako Pure Chemical Corporation (Tokyo, Japan). Clotrimazole was from the Tokyo Chemical Industry (Tokyo, Japan). Gossypol was from LKT Laboratories (St. Paul, MN, USA). SA was purchased from Otsuka Pharmaceutical Co., Ltd (Tokushima, Japan). NTC–CARBOMER 380 for high-viscosity CVP (HV-CVP) and NTC–CARBOMER 381 for CVP (Nikko Chemicals Co. Ltd, Tokyo, Japan) were used as CVPs. Poly acrylic acid (Sigma-Aldrich) was used as non-cross-linked CVP (Ncl-CVP). These CVPs were dissolved in sterile distilled water (Otsuka Pharmaceutical Co., Ltd) and adjusted to pH 7.0 with endotoxin-free sodium hydroxide solution (Nacalai Tesque Ltd, Kyoto, Japan). Pharmaceutical base materials #1–8 and #10–18 were generously provided by Toko Yakuhin Kogyo Co. Ltd (Toyama, Japan) and the compositions of these pharmaceutical base materials are shown in Supplementary data Table 1.
Antibodies for FACS analyses
For studies in mice, single-cell suspension cells were stained with 7-Amino-Actinomycin D (7–AAD) or f LIVE/DEA Fixable Yellow (Thermo Fisher Scientific, Waltham, MA, USA) to identify dead cells. To stain surface antigens, the following antibodies were used: Alexa Fluor700-conjugated anti-CD3 (Biolegend, San Diego, CA, USA; clone: 17A2, phycoerythrin (PE)-cyanine (Cy)7-conjugated anti-CD19 (Biolegend; clone: B4), Brilliant violet 570-conjugated anti-F4/80 (Biolegend; clone: BM8), fluorescein isothiocyanate (FITC)-conjugated anti-IA/IE antibody (Biolegend; clone: M5/114.15.2), allophycocyanin (APC)-conjugated anti-CD11c (Biolegend; clone: N418), Pacific blue-conjugated anti-CD103 (Biolegend; clone: 2E7), APC-Cy7-conjugated anti-CD11b (Biolegend; clone: M1/70), PE-conjugated anti-H–2Kb bound to SIINFEKL (Biolegend; clone: 25-D1.16), Pacific blue-conjugated anti-B220 (Biolegend; clone: RA3–6B2), APC-conjugated anti-CD38 (Biolegend; clone: 90), Brilliant violet 650-conjugated anti-CD69 (Biolegend; clone: H1.2F3), PE-conjugated anti-Fas (Biolegend; clone: SA367H8), FITC-conjugated-GL7 (Biolegend; clone: GL7), Pacific Blue-conjugated anti-CD4 (Biolegend; clone: GK1.5), APC-conjugated anti-PD–1 (Biolegend; clone: 29F.1A12), and (or) FITC-conjugated anti-CXCR5 (Biolegend; clone: L138D7). For staining intracellular cytokines, PE-conjugated anti-Foxp3 (BD Biosciences, San Jose, CA, USA; clone: MF23) was used. For studies in cynomolgus monkeys, single-cell suspensions of PBMC or BALF cells were stained with eBioscience™ Fixable Viability dye eFluor™ 780 (Thermo Fisher Scientific) to identify dead cells. Subsequently, the cells were washed and then stained with APC-conjugated anti-CD3 (Thermo Fisher Scientific; clone: SP34–2) and FITC-conjugated anti-CD4 (Thermo Fisher Scientific; clone: M–T477). Then, the cells were fixed and permeabilized with BD Cytofix/Cytoperm Fixation/Permeabilization Kit (BD Biosciences). For staining intracellular cytokines, cells were incubated with PE-conjugated anti-IL-4 (Invitrogen, Waltham, MA, USA; clone: 8D4–8), PE-conjugated anti-IL-5 (Invitrogen; clone: TRFK5), PE-conjugated anti-IFNγ (Invitrogen; clone: 4S.B3), or PE-conjugated anti-TNFα (Thermo Fisher Scientific; clone: MAB11) antibodies.
Mice and immunizations
Six–to–seven-week-old female BALB/c mice (16–21 g) were obtained from Japan SLC, Inc. (Tokyo, Japan). Mice lacking the type I IFN receptor, Ifnra1˗/˗, generated in a previous study were kindly provided by Dr. Takeshi Kurosu (Department of Virology I, National Institute of Infectious Diseases, Musashi–murayama, Tokyo, Japan) and bred in-house. The mice were housed in rooms maintained at 23 °C ± 1 °C and at 50% ± 10% relative humidity under a 12 h light/12 h dark cycle. The mice were acclimated for at least 3 days before the experiments commenced. The animal experiments were performed according to the guidelines of the Institutional Animal Care and Use Committee of the National Institute of Infectious Diseases, Tokyo, Japan. The study was reviewed and approved by the Institutional Animal Care and Use Committee of the National Institute of Infectious Diseases (approval numbers 117099, 120099, 120100, 120101, 120102, 121196, 122015, 123067, 122146, 121192, and 122147). For immunization, the mice were anesthetized by intraperitoneal injection of 100 mg/10 ml/kg ketamine (Ketalar®; Daiichi–Sankyo Co., Ltd, Tokyo, Japan) and 10 mg/10 ml/kg xylazine (Selactar®; Bayer, Ltd, Tokyo, Japan), then immunized with SV, WPV, or SV with adjuvant intranasally under anesthesia. The administration volume was 15 μl into each nostril. For existing adjuvants, alum (100 µg/mouse)53, poly I:C (10 µg/mouse)54, CpG K3 (50 µg/mouse)55, R848 (10 µg/mouse)56, Hydroxypropyl-β-cyclodextrin (10% w/v)57, DMXAA (300 µg/mouse)35, and AddaVax (12.5%v/v)35 were used. Approved drugs were dissolved in a final concentration of 6.7% of dimethylsulfoxide (DMSO). As a vehicle control, a 6.7% DMSO-administered group was included for some experiments. SV, WPV, and adjuvant concentrations were adjusted by dilution with SA at a dosing volume of 30 μl/mouse. The dosing amounts of SV and WPV were 1 μg protein/mouse, and the dosing amounts of SARS-CoV-2 spike protein S1 and Fc-tag antigen were 3 μg protein/mouse. For lung biomarker gene expression analyses and leucopenic toxicity assessment, according to previous studies, at 16 h after inoculation, mice were euthanized via cervical dislocation under anesthesia (intraperitoneal injection of 100 mg/10 ml/kg ketamine and 10 mg/10 ml/kg xylazine) and then the lungs and blood were collected. For immunizations, 21 days after inoculation, mice were euthanized via cervical dislocation under anesthesia (intraperitoneal injection of 100 mg/10 ml/kg ketamine and 10 mg/10 ml/kg xylazine), and BALF and blood samples were collected. In some experiments, boost vaccination was performed at 21 days after the first immunization. At 14 days after boosting, mice were euthanized via cervical dislocation under anesthesia (intraperitoneal injection of 100 mg/10 ml/kg ketamine and 10 mg/10 ml/kg xylazine), and BALF and blood samples were collected. To evaluate the adjuvanticity of CVP in subcutaneous vaccination, mice were subcutaneously immunized with 3 µg protein/mouse of SV with or without CVP at 200 µl/mouse. Two weeks after subcutaneous inoculation, serum was collected under anesthesia (intraperitoneal injection of 100 mg/10 ml/kg ketamine and 10 mg/10 ml/kg xylazine), and antigen-specific IgG1 and IgG2a antibody production levels were evaluated. For histopathological analyses, 14 days after nasal inoculation, mice were euthanized via cervical dislocation under anesthesia under anesthesia (intraperitoneal injection of 100 mg/10 ml/kg ketamine and 10 mg/10 ml/kg xylazine), and lungs were inflated with, and fixed in, 10% formaldehyde to perform histopathological analyses. In the immunization assay using OVA antigen, mice were nasally administered 50 μg of OVA antigen per mouse, with 25 μl delivered into each nostril under anesthesia (intraperitoneal injection of 100 mg/10 ml/kg ketamine and 10 mg/10 ml/kg xylazine).
Influenza virus infection
Twenty-one days after immunization, the mice were anesthetized by intraperitoneal injection with ketamine (Ketalar®; Daiichi–Sankyo Co., Ltd, Tokyo, Japan, 100 mg/10 ml/kg) and xylazine (Selactar®; Bayer, Ltd, Tokyo, Japan, 10 mg/10 ml/kg) and then intranasally infected with A/California/7/2009 [A(H1N1)pdm09] influenza virus at a dose of 10 × LD50 in a volume of 50 μl/mouse. All mice were monitored daily for survival and body weight loss until 14 days after infection. To assess influenza virus titers, BALF, and nasal wash were collected 3 days after infection under anesthesia (intraperitoneal injection of 100 mg/10 ml/kg ketamine and 10 mg/10 ml/kg xylazine).
Lung biomarker gene expression analyses for screening
For the gene expression analyses of animals treated with influenza vaccine, the existing adjuvant, and pharmaceutical base materials, the lungs were collected for a QuantiGene Plex (QGP) assay. For immunization, the mice were anesthetized by intraperitoneal injection of 100 mg/10 ml/kg ketamine and 10 mg/10 ml/kg xylazine. Briefly, the lung lysates were prepared and the QGP assay was performed, as described in our previous studies33,34,35. The lung specimens were immediately stored in RNAlater (Thermo Fisher Scientific Japan, Kanagawa, Japan) and homogenized before the QGP assay was performed according to the instructions provided with the QuantiGene Plex Reagent System (Panomics/Affymetrix, Fremont, CA, USA), as described previously33,34,35. The probes for the biomarker genes were designed as described previously33,34,35. WPV-like toxicity levels were calculated from the gene expression levels using ordinal logistic regression analysis with JMP 12.01 statistical software (SAS Institute, NC, USA), as previously reported33,34,35.
Measurement of white blood cell (WBC) counts and cytokine levels in sera
The leukopenic toxicity test was performed according to a previously reported method33. The concentrations of the tested vaccine antigens (SV or WPV) were adjusted to 15 μg HA/0.5 ml. Each adjuvant was mixed with SV. The tested vaccine was injected intraperitoneally at a dose of 0.5 ml/mouse. At 16 h after injection, the mice were anesthetized by intraperitoneal injection of 100 mg/10 ml/kg ketamine and 10 mg/10 ml/kg xylazine. Blood samples were collected via the inferior vena cava. The WBC and platelet counts were determined using an automatic hemocytometer, the Celltac MEK–6450 (Nihon Kohden, Tokyo, Japan). Sera were isolated using Capiject (Terumo Medical Corporation, Somerset, NJ, USA). The concentrations of cytokines and chemokines in mice sera were measured using the MILLIPLEX® Mouse Cytokine/Chemokine Magnetic Bead kit (Merck Millipore, Darmstadt, Germany).
Collection of bronchoalveolar lavage fluid (BALF) and nasal wash
The mice were anesthetized by intraperitoneal injection of 100 mg/10 ml/kg ketamine and 10 mg/10 ml/kg xylazine. After cannulating the trachea, the lungs were lavaged with 1% bovine serum albumin (BSA) containing phosphate-buffered saline (PBS) (1.0 ml). The supernatants of the BALF samples were collected after centrifugation at 3000 × g at 4 °C for 15 min. The BALF samples were stored at −80 °C prior to analysis. The extent of lung tissue injury was assessed by measuring the LDH activity in the BALF using the Cytotoxicity LDH Assay Kit–WST (Dojindo, Kumamoto, Japan) in accordance with the manufacturer’s instructions. The head of the mouse was removed, and the lower jaw was excised. A syringe needle was inserted into the posterior opening of the nasopharynx and then 1 ml of PBS containing 0.1% BSA was injected into the opening three times; the outflow was collected as the nasal wash. The nasal wash was centrifuged to remove cellular debris and used for virus titration.
Measurement of the influenza vaccine antigen-specific antibody concentration
SV antigen-specific antibodies in the nasal wash, BALF, and sera samples were determined using an enzyme-linked immunosorbent assay (ELISA). Briefly, 96-well Falcon microtest assay plates (BD Biosciences) were coated with recombinant influenza A H1N1 (A/Puerto Rico/8/34, PR8) hemagglutinin (HA) protein (Sino Biological Inc., Beijing, China) or purified A/New Caledonia/20/99; H1N1 HA protein (Kitasato Institute, Tokyo, Japan) at a 1.0 μg/ml concentration. After blocking (using 1% BSA in PBS), two-fold serial dilutions of the samples or mouse anti-PR8 HA monoclonal antibody (Sino Biological, North Wales, PA, USA), used as a standard, were added and incubated overnight at 4 °C. Horseradish peroxidase-labeled goat anti-mouse IgG1, IgG2a, or IgA antibodies (Southern Biotechnology Associates, Birmingham, AL, USA) were added and the color was allowed to develop for 15 min at room temperature in 100 μl of 1.1 mM 2,2ʹ-azino bis (3-ethylbenz-thiazoline-6-sulfonic acid) (EMD Biosciences, La Jolla, CA, USA). Absorbance (450 nm) was detected using a microplate reader (Synergy LX Multi–Mode Reader, BioTek, Winooski, VT, USA). The antibody concentration was calculated based on a standard curve plotted using data obtained from experiments with mouse anti-PR8 HA monoclonal antibody and recombinant PR8 protein-coated 96-well plates. In particular, the sample was serially diluted and the antibody concentration was determined from the standard curve from a range within which the absorbance and dilution concentration were linear (r2 > 0.95). The concentration of OVA-specific IgE or total IgE antibodies in sera were determined using a LEGEND MAX™ Mouse OVA Specific IgE ELISA Kit or ELISA MAX™ Deluxe Set Mouse IgE Kit (Biolegend) in accordance with the manufacturer’s instructions.
ELISPOT assay
The ELISPOT assay was used to determine mouse IFNγ-producing cells from isolated spleen or lung lymphocytes. IFNγ spot-forming cell assessment was conducted according to the manufacturer’s instructions (ImmunoSpot; Cellular Technology Ltd, Shaker Heights, OH, USA). Briefly, single-cell suspensions of mononuclear cells were seeded in 96-well plates at 1 × 105 cells/well, 1 μM of peptide was added, and the plates were incubated for 24 h (5% CO2, 37 °C). Cytokine ELISPOTs were evaluated using a CTL ImmunoSpot analyzer (Cellular Technology Ltd).
Flow cytometry analyses
For flow cytometry analyses on mice, the mice were anesthetized by intraperitoneal injection with ketamine (100 mg/10 ml/kg) and xylazine (10 mg/10 ml/kg). Immediately before collection, the lungs were perfused with PBS through the apex of the heart before tissue collection, and the lungs were inflated through the bronchi with 1 ml of digestion solution comprising 25 mM HEPES containing RPMI 1640 (Invitrogen) with 5% fetal calf serum (FCS), 10 μg/ml DNase I (Roche, Basel, Switzerland), and 1 mg/ml collagenase IV (Invitrogen). The inflated lung was placed on 2 ml of digestion solution and minced, followed by digestion for 45 min at 37 °C with shaking. The reaction was stopped by placing the sample on ice with 2 mM ethylenediaminetetraacetic acid (EDTA) in solution. Single-cell suspensions of lung tissue were prepared by individually mashing the tissues through a 70 μm cell strainer. The cells were pelleted at 300 × g for 15 min at 4 °C. The supernatant was then removed. The single-cell suspensions were also red blood cells (RBC)-lysed with ammonium chloride solution (StemCell Technologies, Vancouver, Canada) for 1 min at room temperature. The RBC lysates were stopped by adding a 10-fold volume of ice-cold FACS buffer (5% FCS containing PBS). The cells were pelleted at 300 × g for 15 min at 4 °C, then washed twice with ice-cold FACS buffer and suspended in FACS buffer. Immediately after harvesting, the spleens were minced with scissors in a solution of 25 mM HEPES containing RPMI 1640 (Invitrogen) with 5% FCS and 10 μg/ml DNase I (Roche, Basel, Switzerland). Single-cell suspensions were obtained by passing the minced spleens through a 70-μm cell strainer. Subsequent processing, including lysis of red blood cells, followed the same procedure as for lung cells.
Single-cell suspensions were incubated with fluorescently conjugated monoclonal antibodies against mouse antigens for 30 min at 4 °C. Detailed information on the antibodies is provided in the reagent section. The cells were washed twice with ice-cold FACS buffer. To exclude dead cells, the cells were suspended in propidium iodide-containing FACS buffer immediately before analysis. For intracellular staining, the cells were incubated for 4–6 h with 10 mg/ml brefeldin A (Sigma-Aldrich), 10% FCS, and 25 mM HEPES containing RPMI 1640 at 37 °C. The cells were washed with FACS buffer and resuspended in FACS buffer. Fixable viability stain 510 (BD Biosciences) was used for viability staining according to the manufacturer’s instructions. After anti-CD16/CD32 monoclonal antibody (Miltenyi Biotec, Auburn, CA, USA) treatment, the cells were stained with antibodies against cell surface markers in FACS buffer for 30 min at 4 °C. The cells were washed with FACS buffer before fixation and permeabilization using the BD Cytofix/Cytoperm Plus Fixation/Permeabilization Kit (BD Biosciences) according to the manufacturer’s instructions. The cells were stained with PE-conjugated rat anti-mouse Foxp3 (R16–715) monoclonal antibody, washed with PBS, and resuspended in FACS buffer for acquisition. Samples were run on a CytoFLEX Flow Cytometer (Beckman Coulter Inc., Brea, CA, USA) and analyzed using FlowJo software (TreeStar, San Carlos, CA, USA).
In the experiment with cynomolgus macaques, single-cell suspensions were prepared as follows. PBMCs were isolated using lymphocyte mammal cell separation medium. The BALF was centrifuged at 300 × g for 10 min at 4 °C, and the pelleted cells were collected. Each cell type was incubated overnight at 37 °C with 5% CO2 in RPMI 1640 medium supplemented with 10% FBS, containing 2 μg/ml SARS-CoV-2 S1 protein and 0.15 μg/ml brefeldin A for PBMCs, or 5 μg/ml SARS-CoV-2 S1 protein and 0.15 μg/ml brefeldin A for BALF cells. The cells were then subjected to FACS analysis. The antibodies used are listed in the Antibodies section. Intracellular staining was performed using the BD Cytofix/Cytoperm™ Fixation/Permeabilization Solution Kit (BD Biosciences) and antibodies against IFNγ, IFNα, IL-4, or IL-5. Dead cells were identified using eBioscience™ Fixable Viability Dye eFluor™ 780 (Thermo Fisher Scientific). Gating strategies for mouse CD103+ DCs, GC B cells, Tfh cells, or cynomolgus monkey BALF cells and PBMCs are shown in Figs. S3–S5.
In vivo CTL assay
Mice were immunized with various types of vaccine twice with a 3-week interval. Ten days after the final immunization, the mice were subjected to an in vivo CTL assay. Spleen cells (1 × 107 cells/ml) from naïve BALB/c mice were incubated with 1 μM influenza virus HA peptide (IYSTVASSL)- or OVA (257–264)-peptide-containing PBS for 2 h at 37 °C. After two PBS washes, the cells (1 × 107 cells/ml) were labeled with different concentrations of carboxyfluorescein diacetate succinimidyl ester (CFSE) (0.25 or 2.5 μM, Molecular Probes, Eugene, OR, USA) at room temperature for 10 min. The labeling was terminated by adding an equal volume of fetal calf serum, followed by two additional washing steps. Five million cells carrying each peptide were mixed and injected intravenously into the immunized mice. Fourteen hours post-injection, spleens were harvested to prepare single-cell suspensions, and CFSE-positive cells were analyzed by flow cytometry, excluding dead cells via propidium iodide (BD Biosciences) staining. Nucleoprotein-specific killing was calculated as follows: specific killing (%) = {1 ˗ [(number of HA (533–541)-carrying cells in immunized mice (CFSE high))/(number of OVA (257–264)-carrying cells in immunized mice (CFSE low))]/[(number of HA (533–541)-carrying cells in normal mice (CFSE high))/(number of OVA (257–264)-carrying cells in normal mice (CFSE low))]} × 100.
Measurement of recombinant SARS-CoV-2 S1 antigen or RBD-specific antibody titers
SARS-CoV-2 S1 antigen or RBD-specific antibody titers were determined. Briefly, 96-well plates were coated with 1 μg/ml HEK293K cell-expressed recombinant SARS-CoV-2 S1 protein with a His-tag, SARS-CoV-2 Spike RBD (XBB.1.5) with a His-tag, or SARS-CoV-2 Spike RBD (JN.1) with a His-tag (ACROBio systems, Newark, DE, USA) overnight at 4 °C. Plates were washed once with wash buffer [PBS containing 0.05% (v/v) polysorbate 20] and then blocked with 5% (w/v) skim milk dissolved in wash buffer for 1 h at 37 °C. Plates were then washed four times and incubated with serially diluted mouse sera for 1 h at 37 °C. Next, plates were washed five times and incubated with goat anti-mouse IgA/IgG1/IgG2a HRP-conjugated secondary antibodies (Thermo Fisher Scientific) for 1 h at 37 °C. Following five additional washes, 1.1 mM 2,2ʹ-azino bis (3-ethylbenz-thiazoline-6-sulfonic acid) was added. The plate was incubated at room temperature in the dark for 10 min, and reactions were stopped by the addition of 2 N H2SO4. Absorbance (450 nm) was detected using a microplate reader (Synergy LX Multi–Mode Reader, BioTek, Winooski, VT, USA). Antibody titers were defined by endpoint dilution with a cut-off signal of OD450 = 0.1. Sera samples that did not produce an optical density of more than 0.1 at 1:100 were determined as 100. BALF and nasal wash samples that did not produce an optical density of more than 0.1 at 1:2 were determined as 2.
SARS-CoV-2 pseudovirus preparation, titration, and neutralization assays
Plasma neutralizing titers were measured with pseudotyped VSVs prepared as previously described17. The pseudotyped VSVs, which encode luciferase, expressing spike antigens derived from SARS-CoV-2 Wuhan (WK–521), XBB.1.5 (the Wuhan spike with T19I, L24S, del25/27, V83A, G142D, del144, H146Q, Q183E, V213E, G252V, G339H, R346T, L368I, S371F, S373P, S375F, T376A, D405N, R408S, K417N, N440K, V445P, G446S, N460K, S477N, T478K, E484A, F486P, F490S, Q498R, N501Y, Y505H, D614G, H655Y, N679K, P681H, N764K, D796Y, Q954H, and N969K mutations), or JN.1 (the Wuhan spike with T19I, R21T, del24/26, A27S, S50L, del69/70, V127F, G142D, del144, F157S, R158G, del211, L212I, V213G, L216F, H245N, A264D, I332V, G339H, K356T, S371F, S373P, S375F, T376A, R403K, D405N, R408S, K417N, N440K, V445H, G446S, N450D, L452W, L455S, N460K, S477N, T478K, N481K, del483, E484K, F486P, Q498R, N501Y, Y505H, E554K, A570V, D614G, P621S, H655Y, N679K, P681R, N764K, D796Y, S939F, Q954H, N969K, and P1143L mutations), were mixed with plasma samples serially diluted in Dulbecco’s modified Eagle’s medium (DMEM) (Fujifilm) supplemented with 2% heat-inactivated FBS (Thermo Fisher Scientific) and 100 U/ml penicillin/streptomycin (Thermo Fisher Scientific). The mixed plasma and viruses were incubated at 37 °C and 5% CO2 in a humid atmosphere for 1 h. Then, the mixture was inoculated onto VeroE6/Tmprss2 cells followed by incubation at 37 °C and 5% CO2 in a humid atmosphere for 24 h. After incubation, the luciferase activity of the cells was measured with Bright–Glo (Promega, Madison, WI, USA) and the GloMax Navigator Microplate Luminometer (Promega). Half-maximal inhibitory concentrations (IC50) were calculated using Prism 9 (GraphPad, San Diego, CA, USA).
Human PBMC study
PBMCs obtained from three healthy adult subjects were purchased from AllCells (Alameda, CA, USA). PBMCs were seeded at a density of 5 × 104 cells per well in a 96-well plate. The culture medium was RPMI 1640 with 10% FCS, 100 U/ml penicillin, and 100 μg/ml streptomycin (Sigma-Aldrich). The cells were cultured at 37 °C with 5% CO2 for 24 h in media containing the following adjuvant concentrations: 10 μg/ml WPV, 100 μg/ml alum, 20 μg/ml R848, 20 μg/ml poly I:C, 5% AddaVax, 0.03% or 0.3% CVP, and 50 μg/ml CpG ODN 2006. Post incubation, culture supernatants were centrifuged at 3000 × g for 10 min at 4 °C, and the supernatants were collected for cytokine analysis. The concentration of cytokines and chemokines in supernatants were measured using the MILLIPLEX MAP Human Cytokine/Chemokine Magnetic Bead Panel Immunology Multiplex Assay (Merck Millipore).
TLR ligand screening with HEK-Blue TLR cells
HEK-Blue human TLR cells (InvivoGen) are NF-κB-secreting alkaline phosphatase reporter cells that stably express mouse TLR genes (Tlr2, 3, 4, 5, 7, 8, or 9). HEK-Blue STAT6–STING–R232 cells (InvivoGen) are STAT6-inducible secreted embryonic alkaline phosphatase (SEAP) reporters that stably overexpress the most prevalent human (h)STING variant R232. TLR or STING stimulation was tested by assessing NF-κB activation in HEK293 cells expressing a given TLR. After incubation for 24 h with the test substance, the optical density (OD) was measured at 650 nm. The following substances were used as positive controls, for TLR2: heat-killed Listeria monocytogenes at 108 cells/ml; for TLR3: poly I:C at 1 μg/ml; for TLR4: E. coli K12 LPS at 100 ng/ml; for TLR5: flagellin at 100 ng/ml; for TLR7: CL097 at 1 μg/ml; for TLR8: CL075 at 10 μg/ml + poly(dT) at 10 μM; for TLR9: CpG ODN 1806 at 1 μg/ml; and for STING: 2ʹ3ʹ-cGAMP at 30 μg/ml. CVP was added at 0.3% concentration. Assays were performed according to the manufacturer’s instructions.
Measurement of influenza virus titers
Virus titers in lung homogenates were measured 3 days post-challenge. As previously described4, virus titers were determined by a 50% tissue culture infectious dose (TCID50) assay. Confluent monolayers of MDCK cells in 96-well plates were inoculated with serial half-log dilutions of virus samples and incubated for 6 days at 34 °C in 5% CO2 in the presence of 10 μg/ml acetylated trypsin (Sigma-Aldrich). Virus titers were calculated for each sample using the Reed–Muench method, based on the cytopathic effects in individual wells observed under an inverted microscope.
SARS-CoV-2 infection
For virus challenge studies, vaccinated mice were intranasally inoculated under anesthesia by intraperitoneal injection with ketamine (100 mg/10 ml/kg) and xylazine (10 mg/10 ml/kg) with the mouse-passaged D614G strain QHmusX strain (1 × TCID50/5 µl for virus RNA levels measurement; 10 × TCID50/50 µl for survival analyses) at 2 weeks post-boost vaccination. The infected mice were monitored for clinical signs of infection and their body weight was measured daily for 14 days or until progression to a humane endpoint. The humane endpoint was defined as the appearance of clinically diagnostic signs of respiratory stress, including respiratory distress, and weight loss of more than 25%. Total RNA was extracted from BALF and nasal washes and was subjected to real-time RT–PCR for viral RNA quantitation of the SARS-CoV-2 genome using a TRIzol™ Plus RNA Purification Kit (Thermo Fisher Scientific) as previously described53. The RNAs were also subjected to real-time RT–PCR for measurement of viral subgenomic RNA (sgRNA) levels using the following primers: SARS2–LeaderF60 (5ʹ–CGATCTCTTGTAGATCTGTTCTCT–3ʹ), SARS2–N28354R (5ʹ–TCTGAGGGTCCACCAAACGT–3ʹ), and SARS2–N28313Fam (FAM–TCAGCGAAATGCACCCCGCA–TAMRA). Viral RNA or sgRNA levels less than 3 × 103 copies/swab were undetectable.
Histopathological analyses
Fourteen days after intranasal administration of SA, 100 µg of alum, or 0.3% CVP, the mice were anesthetized by intraperitoneal injection with ketamine (100 mg/10 ml/kg) and xylazine (10 mg/10 ml/kg), and then lungs were inflated with and fixed in 4% paraformaldehyde. To prepare paraffin sections, the tissues were gradually dehydrated and embedded in paraffin. The sections (5 µm) were prepared and stained with periodic acid–Schiff (PAS) using the Periodic Acid–Schiff Kit (Sigma-Aldrich). Images were acquired by an Olympus BX53 optical microscope (Olympus, Tokyo, Japan).
Cynomolgus monkeys
Female and male Cambodia cynomolgus monkeys (Macaca fascicularis) imported from China were purchased and experiments were conducted in Hamri Co., Ltd (Ibaraki, Japan). The animals were purpose-bred and negative for anti-herpes simian B virus (Macacine herpesvirus 1) antibody, anti-simian varicella virus antibody, tuberculin, Salmonella, Shigella, and anti-SARS-CoV-2 neutralizing antibody. The monkeys were 2 years old and weighed 2.2–4.0 kg at initiation. The monkeys were individually housed in paired cages and maintained under controlled environmental conditions with a 12 h light/dark cycle. All animal care and manipulations were performed at Hamri Co., Ltd. All animal experiments were approved by the Hamri Co., Ltd Institutional Animal Care and Use Committee (Approval No. 22–015) and performed in accordance with the animal welfare bylaws of Hamri Co., Ltd, which is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International.
Immunization of cynomolgus monkeys
The overall study design is shown in Fig. 8A. Body temperature was measured with a small implantable thermo logger (DST nano–T; Star–Oddi) that was set intraperitoneally under ketamine anesthesia (10 mg/0.2 ml/kg ketamine and medetomidine 50 μg/0.2 ml/kg by intramuscular injection) at least 5 days prior to vaccination. To minimize body weight bias among the groups, the animals were assigned according to body weight. Each of the experimental groups included four cynomolgus monkeys. For intranasal immunization, the animals were nasally inoculated with 50 μg/250 μl of the SARS-CoV-2 (Wuhan strain) spike trimer in each nostril (total 100 μg/body) under anesthesia (10 mg/0.2 ml/kg ketamine and medetomidine 50 μg/0.2 ml/kg by intramuscular injection). For vaccines containing CVP, a solution of 50 μg/250 μl SARS-CoV-2 spike trimer with 0.375% CVP was administered in each nostril (total 100 μg/body). Anesthesia was induced with an intramuscular injection of ketamine hydrochloride (10 mg/0.2 ml/kg by intramuscular injection), supplemented with inhaled isoflurane as needed. The nasal inoculation was performed using a specialized device kindly provided by Toko Yakuhin Kogyo Co., Ltd (Toyama, Japan), with the angle adjusted to ensure the solution covered the entire mucosal surface by lifting the nose.
For intramuscular injection, 25 μg/500 μl of the SARS-CoV-2 (Wuhan strain) spike trimer vaccine, with or without 0.37% CVP, was administered under anesthesia (10 mg/0.2 ml/kg ketamine and medetomidine 50 μg/0.2 ml/kg by intramuscular injection) using a syringe and needle into one thigh opposite the induction site. The all monkeys were sacrificed by exsanguination under excess ketamine anesthesia (20 mg/0.2 ml/kg ketamine and medetomidine 100 μg/0.2 ml/kg by intramuscular injection) at 77 days post priming and then necropsied.
Blood collection of cynomolgus monkeys
Blood sampling was performed under anesthesia with ketamine hydrochloride (10 mg/0.2 ml/kg by intramuscular injection), using a syringe and needle to draw approximately 1 ml of blood from the radial or femoral vein. The blood was transferred to Sepaclean A tubes (Eiken Chemical Co., Ltd, Tokyo, Japan), allowed to coagulate at room temperature for 15–30 min, then centrifuged (2300 × g, 10 min, 4 °C). The serum was aliquoted into two 200 μl portions, frozen on dry ice, and stored at −70 °C.
Nasal wash and BALF collection from cynomolgus monkeys
Nasal wash fluid was collected as follows. Under ketamine hydrochloride anesthesia (5–10 mg/0.1–0.2 ml/kg by intramuscular injection), an appropriate amount of lidocaine was sprayed around the pharynx. An animal probe with a bent tip was inserted orally and directed from the oropharynx to the nasopharynx. Approximately 2 ml of PBS (pH 7.4) was flushed through the probe, and the wash fluid flowing out of the nostril was collected in a 5 ml tube. The collected fluid was centrifuged (2300 × g, 10 min, 24 °C), and the supernatant was aliquoted into 2 ml tubes, frozen on dry ice, and stored at −70 °C.
BALF was collected under anesthesia with ketamine hydrochloride (10 mg/0.2 ml/kg by intramuscular injection), with lidocaine sprayed around the larynx. After confirming the airway with a laryngoscope, a tracheal tube was inserted. A feeding catheter was passed through the tracheal tube, and approximately 10 ml of PBS (pH 7.4) was slowly infused. The PBS was gently withdrawn with a syringe and the process was repeated to collect the lavage fluid. The collected fluid was centrifuged (2300 × g, 10 min, 24 °C), and the supernatant was aliquoted into 15 ml tubes, frozen on dry ice, and stored at −70 °C.
Neutralizing antibody titer testing
Cynomolgus monkey sera were analyzed for neutralizing antibody levels using SARS-CoV-2 (WK–521) and TMPRSS–2 expressing Vero E6 cells. DMEM (Thermo Fisher Scientific) supplemented with 2% heat-inactivated FBS and 1 mg/ml gentamicin was used as the assay medium. The serum samples from the vaccinated cynomolgus monkeys were heat-inactivated at 56 °C for approximately 30 min to remove non-specific inhibitors prior to neutralizing antibody titer testing. The vaccinated sera were two-fold serially diluted over a range covering the neutralizing window of all samples. A 100-µl aliquot of each sample was mixed with an equal volume of WK–521 suspensions in a 96-well plate, and 100 µl of each mixture was then added to individual culture-plate wells. The plates were incubated for approximately 1 h at room temperature to allow for neutralization. Then, 3 × 104 TMPRSS–2-expressing Vero E6 cells in 100 µl of assay medium were added to each well (100 TCID50/well), followed by incubation at 37 °C with 5% CO2 for 3 days. The assay was performed in duplicate for each sample. Living cells were examined using CellTiter–Glo 2.0 (G9243; Promega) according to the manufacturer’s instructions. Briefly, 100 μl of supernatant was removed from each well and replaced with 100 μl of CellTiter–Glo 2.0 solution. The plates were incubated at room temperature for approximately 30 min under light-shielding conditions. Then, 100 μl of the mixtures were transferred to 96-well plates and the luminescence intensity was measured using an EnSpire 2300 Multilabel Plate Reader (PerkinElmer, Waltham, MA, USA). The neutralizing antibody titer in the serum samples was calculated as the reciprocal of the highest dilution at which the percentage of viable cells was ≥ 50%. The percentage of viable cells was calculated according to the following formula:
where VC indicates the virus control and CC indicates the cell control. When the titer was < 50% of the viable cells, the titer was set as the reciprocal of half of the first dilution value.
Statistical analyses
Statistical analysis was performed using GraphPad Prism 6.0 software (GraphPad Software, La Jolla, CA, USA). An unpaired Student’s t-test was used to compare the means of two groups. The means of three or more groups, with one variable, were compared using one-way ANOVA. Where significant differences were observed, Dunnett’s multiple comparison test was used to identify differences between the control group and another group with an unequal number of samples. Data with p < 0.05 were considered statistically significant. Graphs of results are shown as the mean ± SEM of untransformed data using GraphPad Prism 6.0 software (GraphPad Software).





Responses