CALRins5-mediated clonal hematopoiesis causes severe hemolytic anemia in a female PGK1Ser320Asn carrier
Introduction
Hereditary hemolytic anemias (HHA) are rare inherited red blood cell (RBC) disorders caused by genetic abnormalities (hemoglobino/membrano/enzymopathies), characterized by anemia due to premature RBC destruction and intrinsic RBC defects. One of the rarer enzymopathies involves a deficiency in phosphoglycerate kinase 1 (PGK1), an X-linked gene critical for ATP production via glycolysis [1]. PGK1 mutations causing deficiency (OMIM#300653) [2] follow an X-linked recessive inheritance pattern, affecting hemizygous males, while female carriers are generally asymptomatic or may have mild symptoms -consistent with mosaicism for PGK1 activity-[2].
Despite PGK1 is ubiquitously expressed, its deficiency mainly affects the blood, central nervous system (CNS), and skeletal muscle, resulting in chronic hemolysis -with or without anemia-, neurological disorders, or myopathies [1]. Likely explained by the causative PGK1 mutation [3], hemolytic anemias (HA) tends to co-occur with CNS defects, whereas myopathies are almost exclusively observed [3, 4].
To date, approximately 40 patients harboring 30 different mutations have been reported [5]. This report presents the first case of severe HA in a female PGK1 mutation heterozygous carrier [6], which became apparent coinciding with the diagnosis of essential thrombocythemia (ET); and elucidates the mechanism by which a clonal disorder transforms a germline recessive disease into a tissue-specific dominant condition.
Methods
Written informed consent was obtained from the participants. Clinical and laboratory data were extracted from electronic records. Details of collection and processing of peripheral blood (PB) samples, histological studies, enzymatic activity, sequencing, gene expression, X-chromosome inactivation (XCI), methylation variant calling, and phasing, can be found in the Supplementary Material.
Results
Case presentation
In June 2021, a 32-year-old woman was admitted to the Emergency Department with a two-week history of asthenia and jaundice without choluria, recent infections, or medications. Laboratory tests identified mild regenerative macrocytic anemia (hemoglobin 10.4 g/dL; mean corpuscular volume (MCV) 105 fL [normal range (NR): 79–99 fL]; reticulocytes count, 204000/µL [NR: 15000–80000/µL]) associated with hemolysis pattern (total and indirect bilirubin (6.4 and 6.1 mg/dL, respectively) [NR: 0.30–1.20 and 0.10–0.30 mg/dL, respectively], LDH 674 U/L [NR: 208–378 U/L], undetectable haptoglobin), thrombocytosis (platelets 814000/µL), along with iron overload (ferritin 176 ng/mL [NR:10–120 ng/mL]) and hypergammaglobulinemia (1.9 g/dL) [NR: 0.8–1.4 g/dL]. Chronic liver disease, monoclonal gammopathy, hereditary spherocytosis, β-thalassemia, and autoimmune origin were excluded. Abdominal ultrasound did not show hepatosplenomegaly.
A PB smear showed anisocytosis, polychromasia, and thrombocytosis with anisothrombia (Fig. S1). Glucose-6-phosphate dehydrogenase (G6PD) and pyruvate kinase (PK) levels were within reference limits.
Throughout recent years (2017–2019), compensated hemolysis was evidenced by normal hemoglobin levels but elevated bilirubin and LDH. In fact, due to recurrent hepatic colic, she underwent cholecystectomy in 2018. Notably, the onset of anemia coincided with the appearance of thrombocytosis (June 2019, Fig. 1A). Her pediatric history indicated she was born after two first-trimester miscarriages and was the oldest of three siblings (Fig. 1B). She suffered from neonatal sepsis caused by coagulase-positive staphylococci, followed by anemia and hyperbilirubinemia requiring RBC transfusion. Despite these early complications, she showed compensated hemolysis without signs of other hematological abnormalities.
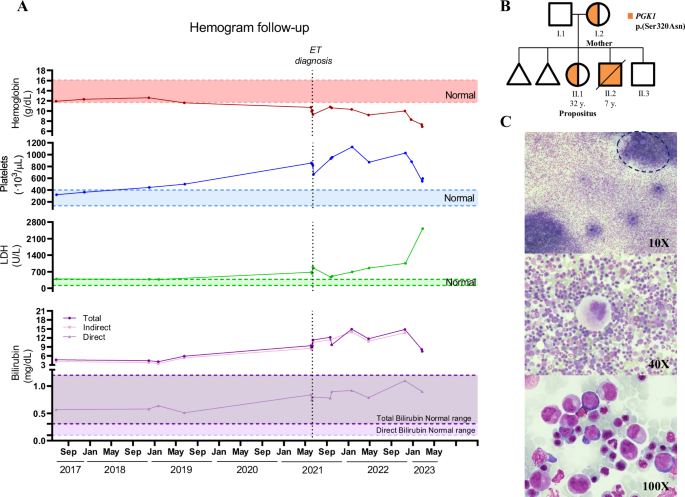
A Hemoglobin (g/dL), platelets (103/µL), LDH (U/L), and bilirubin (mg/dL) levels of the propositus are shown over a 6-year period (June/2017–February/2023). The time of diagnosis of essential thrombocythemia (ET) is indicated. B Pedigree showing the presence of the PGK1 p.Ser320Asn variant in family members. C Bone marrow aspirate of the propositus (II.1) at admission showing marked hypercellularity with hyperplasia of megakaryocytes (top), characteristically enlarged, mature, with hyperlobulated nuclei (middle), along with erythroid hyperplasia (bottom). The dashed line indicates an enriched zone in megakaryocytes within the cell clump.
The patient’s family history revealed that whereas her youngest brother was healthy, her middle brother suffered severe HA from the neonatal period, with blood transfusions every 2–3 weeks, and spastic quadriplegia. Despite splenectomy was performed, he died in 2000 (at 7-year-old) of severe encephalopathy and HA. A post-mortem familial genetic study revealed the first case of PGK-Murcia mutation (PGK1Ser320Asn) [6]. Then, his mother and sister (our patient at 16 year-old) were identified as asymptomatic female carriers [6].
Following clinical protocols, a customized sequencing panel for HHA confirmed the only presence of the PGK1Ser320Asn mutation (Table S1), while other potential causes of jaundice were discarded. Evaluation of persistent thrombocytosis by bone marrow (BM) aspirate (Fig. 1C) and biopsy (Fig. S2) revealed marked myeloid hyperplasia of the erythroid and megakaryocytic lineages (with enlarged, mature, and hyperlobulated megakaryocytes). After ruling out JAK2 mutations, BCR::ABL transcripts, or cytogenetic abnormalities, sequencing studies for myeloid malignancies were performed revealing a CALRins5 mutation (type II) with a variant allele frequency (VAF) of 28.1%. Altogether, the propositus was diagnosed with CALR-mutated ET and HHA due to PGK1 deficiency, which is currently transfusion-dependent.
Biological and molecular studies
The activity quantification of enzymopathies-associated enzymes showed normality for all tested enzymes in the mother, while G6PD, hexokinase (HK), and PK activities were elevated in the propositus (Fig. 2A). Contrarily, PGK1 activity, near the lower limit of the normal range, together with severe reticulocytosis (480000/µL), supported a PGK1 deficiency. Additionally, erythrocyte deformability, measured by the laser-assisted optical rotation cell analyzer (LoRRca) Osmoscan module, showed a normal Osmotic Gradient Ektacytometry curve in the mother but slightly right-shifted curve in the patient, suggesting moderate overhydration likely due to metabolic impairment (Fig. 2B). Similarly to other erythroenzymopathies, the results of the rheological parameters were within the normal range (Table S2).
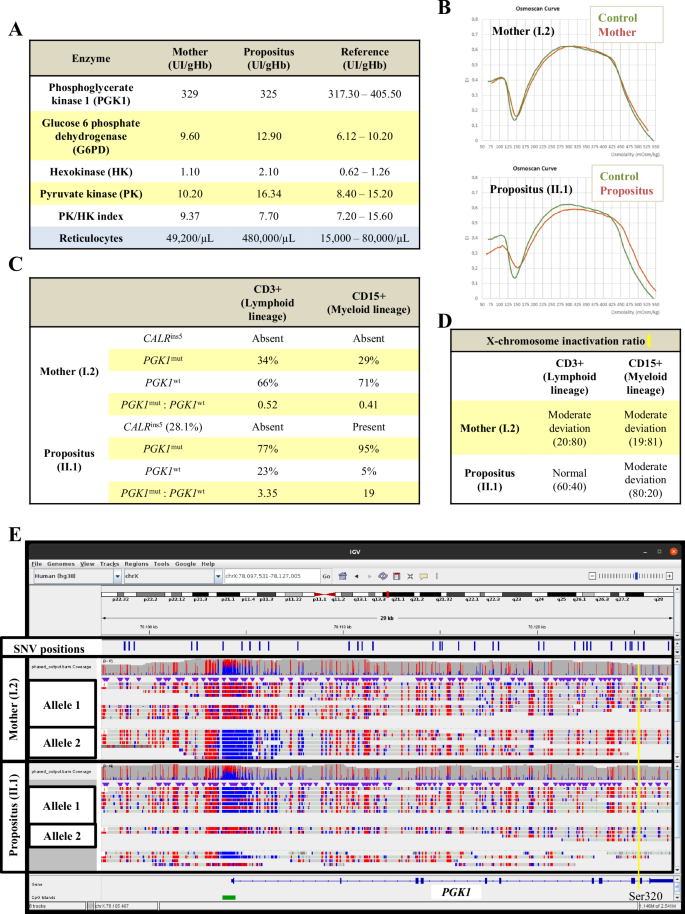
A Enzymopathies-related enzyme activity levels of phosphoglycerate kinase 1 (PGK1), glucose-6-phosphate dehydrogenase (G6PD), hexokinase (HK), and pyruvate kinase (PK) were quantified. This table also shows the number of reticulocytes in the propositus (II.1) and her mother (I.2). Note that the PGK1 activity in the propositus (near the lower limit of the normal range), together with the severe reticulocytosis, supports a PGK1 deficiency. B Osmoscan profiles in the mother (I.2) and the propositus (II.1) using a Laser-Assisted Optical Rotation Cell Analyzer (LoRRca). The osmotic gradient ektacytometry (OGE) reflects the deformability of erythrocytes measured in a suspension medium with constant shear stress and a variable osmolality. Informative parameters of this curve are shown in Table S2. C CALRins5 identification, quantification of VAF and mRNA levels of mutated and wild-type PGK1 alleles in the CD3+ and CD15 + PB population of the mother (I.2) and the propositus (II.1). D X-chromosome inactivation ratio (269:279) by HUMARA assay in CD3+ and CD15 + PB maternal (I.2) and propositus (II.1) populations. The following reference limits were established: normal, <80:20; moderate deviation, 80:20–90:10; and marked deviation, >90:10. E Methylation studies of unmanipulated genomic DNA from myeloid progenitors. The CpG island of the PGK1 promoter is shown in green. Methylated bases are shown in red, while unmethylated bases are shown in blue. Single nucleotide variant (SNV) positions are marked in blue. The yellow line indicates the position of the PGK1 p.Ser320Asn variant. The two alleles differentiated by phasing are indicated. PGK1 p.Ser320Asn variant is detected in allele 1.
Next, we evaluated whether the somatic CALR mutation expanded hematopoietic progenitors with PGK1Ser320Asn. As expected, the CALRins5 mutation was identified exclusively in CD15+ (but not in CD3+ cells or in hair follicles) from the propositus (Fig. 2C). Then, PGK1 mutated (PGK1mut) and wild-type (PGK1wt) mRNA levels were evaluated in both female carriers. Whereas in the mother the PGK1mut:PGK1wt ratio was <0.6 in CD3+ and CD15+ cells, supporting the predominant expression of the PGK1wt form; in the propositus, the ratios were greater than 1 for both CD3+ and CD15+ cells, being even notably higher in the myeloid population (3.35 vs 19), confirming the predominant expression of the mutated form particularly in the myeloid line (Fig. 2C).
After discarding mutations in the Xist gene, we performed the HUMARA assay in CD3+ and CD15+ cells from both carriers. Consistent with previous results, the XCI allele ratios (269:279) showed moderate skewing in CD3+ and CD15+ cells from the mother (20:80 and 19:81, respectively) (Fig. 1D). In the propositus, however, while the ratio was normal for CD3+ cells (60:40), it was moderately skewed for CD15+ cells, but in the opposite direction to that observed in her mother (80:20), supporting that the most inactivated allele in the maternal myeloid cells was the most activated in the propositus’ CD15+ cells.
Finally, nanopore sequencing of unmanipulated genomic DNA from maternal and propositus CD15+ cells allowed assessing in a single approach the methylation status of the CpG island of the PGK1 promoter and the entire gene sequence for each allele. Consistent with mRNA analysis, the mother had methylated the PGK1mut promoter, while the PGK1wt was predominantly unmethylated (Fig. 2E). Conversely, in the propositus, the PGK1mut allele was predominantly unmethylated, while the PGK1wt allele was methylated.
Discussion
Epigenetics has revolutionized the classical concepts of dominant and recessive X-linked inheritance [7]. In 1961, Mary Lyon proposed the random and maintained inactivation of one of the two X-chromosomes in female cells during early development [7]. Today, it is known that up to 20–30% of healthy women may have highly skewed inactivation ratios, affecting the clinical expression of X-linked diseases in female carriers, ranging from normal expression to complete expression of the defect [7,8,9]. It has been proposed that this phenomenon could be a protective mechanism against X-linked disorders [7], as in an F8 and PGK1 mutated carrier [10], or in the propositus’ mother (with moderate XCI and normal PGK1 activity). Our results showed that skewed inactivation can also be driven by an acquired oncogenic event, which expands an epigenetic signature in a single tissue with severe clinical consequences. Thus, the propositus and her mother were carriers for the PGK1-Murcia variant, which caused severe HA and encephalopathy in her younger brother [6]. When the family was firstly described, both heterozygous females showed normal PGK1 activity [6]. Over time, however, the propositus developed compensated chronic hemolysis without anemia, but, in adulthood, coinciding with the onset of progressive thrombocytosis, she manifested anemia. Despite high reticulocyte counts, PGK1 activity decreased, and erythrocytes showed an altered Osmoscan profile, similar to that observed in other enzymopathies [11]. However, since this is the first time that an Osmoscan profile of PGK deficiency has been evaluated, further studies are needed to confirm the specificity of this associated profile.
A relevance finding is the dissociation of HA from neurological impairment in the propositus, unlike her brother [6], likely explained by the different degrees of XCI in specific tissues [7,8,9]. Accordingly, the HUMARA assay revealed moderately skewed inactivation in the propositus’ myeloid (but not lymphoid) progenitors. In line with our results, skewed XCI in the substantia nigra has been linked to parkinsonism in PGK1mut carriers [2].
Nanopore sequencing confirmed the allele-specific differential methylation of the PGK1 promoter associated with the presence of the mutated CALR clone, supplying the strengths of nanopore sequencing in both germline and oncogenic disorders. The methylation profiles led to different expression levels of PGK1mut and PGK1wt, explaining the dramatic clinical differences between both female carriers, despite their shared genotype.
Unfortunately, beyond the potential benefits of splenectomy or allogeneic BM transplantation [6], there is no available therapy for PGK1 deficiency. Our findings support that targeted therapies [12], as the potential use of CRISPR/Cas9 demethylation methods [13], could be promising. Particularly, therapies efficiently reducing the CALRins5 allele burden, such as peginterferon, bomedemstat or the new anti-CALR monoclonal antibodies [14, 15], could ameliorate anemia symptoms.
In conclusion, our study demonstrated how a late somatic oncogenic event (CALRins5) [16], expanded the epigenetic signature of the oncogenic CD15+ clone (unmethylated promoter for PGK1Ser320Asn allele), transforming a recessive X-linked disorder into a dominant one in a specific tissue, causing anemia without the expected neurological impairment [6].
Responses