Convergent evolution of complex adaptive traits modulates angiogenesis in high-altitude Andean and Himalayan human populations
Introduction
Convergent evolution refers to an evolutionary scenario in which different species or populations independently develop the same (or similar) biological trait instead of inheriting it from a common ancestor. In particular, convergent adaptation occurs when distantly related species/populations, which live in distinct geographical areas or epochs, occupy comparable ecological settings and are subjected to the same selective pressures, thus being similarly targeted by natural selection1. Several cases of convergent evolution have been described so far in a number of animal and plant taxa1,2,3,4, with fewer examples (e.g., skin pigmentation5, malaria resistance6, lactose tolerance7 and metabolic adaptations to cold climates7) being instead reported in the human species when comparing adaptive traits among populations of different ancestry. Nevertheless, experimental approaches to accurately characterise genomic variability within and between species/populations, as well as inferential statistical methods suitable to investigate how far phenotypic convergence is achieved by means of genetic convergence, have been only recently conceived. This is especially the case of complex (i.e., polygenic) adaptive traits, whose evolution is regulated by changes at several genes that are functionally related and contribute to the modulation of the same biological function8. Development of methods able to test the occurrence of selective events under a realistic approximation of a polygenic adaptation model has thus opened new possibilities to assess whether natural selection acted on the same genetic variants or genes or functional pathways in species/populations showing shared or similar adaptive traits9.
To this end, human adaptation to the high-altitude environment represents a valuable case study because the main selective pressure acting on populations dwelling at altitude (i.e., hypobaric hypoxia) cannot be mitigated by cultural adaptations and thus acts with the same intensity on human groups living at comparable altitudes irrespectively to their ancestry, geographical locations and socio-cultural contexts. We can thus hypothesise that Himalayan and Andean high-altitude populations, which represent the main human groups whose ancestors have had to cope with such a selective pressure, experienced convergent evolution at least to some extent. Despite this assumption, limited overlapping of potential adaptive loci (e.g., entailing variants at the EGLN1, EDNRA and EPAS1 genes) has been described so far between them10,11,12,13. This may be partially due to the limitations imposed by studies that to date focused mainly on selective sweeps. Indeed, these adaptive mechanisms are mediated by the action of natural selection on single/few genetic variants at the same gene, which implies that these loci can exert a large effect on a given biological trait8. That being so, selective sweeps have low probability to occur in human populations, who have long maintained low effective population sizes and have accumulated low genetic variability and thus need several thousands of years to evolve14. Coupled with the considerably more recent human colonisation of the high-altitude ecological niche in the Andes than in the Himalayas, as supported by estimates at around 9 thousand years ago (kya) for the divergence between Andean and lowland South American populations15 and by archaeological evidence for occupation of the Tibetan Plateau since Palaeolithic times16, this suggests selective sweeps might be not the predominant drivers of adaptation of Andean people to such a challenging environment.
Consistent with this scenario, Andeans indeed evolved less effective genetically regulated physiological adjustments than populations of Tibetan/Sherpa ancestry, especially to counteract long-term detrimental effects of hypobaric hypoxia17,18. When compared with acclimatized Native American lowlanders, they exhibit traits contributing to enhanced efficiency of respiratory functions and oxygen utilisation, such as larger lung volumes and chest dimensions, narrower alveolar to arterial oxygen gradients, less hypoxia-induced pulmonary vasoconstriction, greater uterine artery blood flow during pregnancy, less altitude-related decrement in maximal exercise oxygen consumption and increased cardiac oxygen utilisation17,18. However, when genetic determinants of these physiological characteristics have been searched for, most of the inferred selective sweeps failed to be replicated by multiple studies, suggesting that their relationships with the observed adaptive traits are far from being fully elucidated15,18,19,20,21.
Results and discussion
To test whether polygenic adaptations played a role in shaping the Andean adaptive phenotype, and to extend to complex traits the investigation of the genetic bases of potential convergent evolution between Andean and Himalayan populations, we assembled a dataset representative of genomic variation at several high-altitude ethnic groups from the Andean region (Supplementary Table 1). After stringent quality control (QC) filtering (see ‘Methods’), 7,966,198 Single Nucleotide Variants (SNVs) from 24 Bolivian Aymara whole genome sequences (WGS)15 were used to perform multiple selection scans (i.e., H12, nSL and LASSI) that collectively are able to test for the occurrence of a range of selective events (see ‘Methods’ for further details concerning the rationale behind the choice of these inferential approaches). The obtained results then informed gene-network analyses aimed at identifying sets of functionally related loci enriched for signatures of natural selection. Furthermore, we applied the same pipeline of analyses on: (i) imputed genotype data for 8,024,216 SNVs from 130 individuals belonging to additional Bolivian Aymara groups, as well as from Aymara, Quechua, and Uros high-altitude Peruvian populations22,23 used as validation datasets and (ii) a control dataset including 24 individuals from three different Amazonian low-altitude groups (i.e., Ashaninka, Cashibo, and Shipibo)23 to filter out adaptive loci shared between high- and low-altitude South American populations, with the aim of focusing exclusively on those typical of Andean peoples.
Identification of Native American low-altitude control populations
Setting the considered Bolivian Aymara WGS into the genomic landscape of indigenous groups from Central and South America enabled us to confirm them as well representative of the genetic variation observable across Andean high-altitude populations (Fig. 1a, Supplementary Information). Comparable results were obtained also when considering imputed data for Andeans, thus providing evidence for consistency between non-imputed and imputed datasets (Supplementary Fig. 1). Such a consistency was further attested by the high and significant correlation observed between pre- and post-imputation identity-by-state (IBS) matrices of individual pairwise differences in the genome-wide proportion of shared alleles (Mantel correlation = 0.95, P < 10−4).

a PCA representing PC1 versus PC2 computed for the 43 un-admixed Native American groups reported in the legend at the bottom right of the plot. Individuals are colour-coded according to their country of origin as reported in the bottom-left legend. The box at the top of the plot details the position of Bolivian Aymara WGS in the considered genetic space with respect to the other high-altitude Andean populations. b Heat map of values for the outgroup-f3 statistic representing the estimated amount of shared genetic drift between the Andean population cluster (marked by a star-like symbol) and each of the considered populations from Central and Southern America (indicated in their approximate geographic locations) A gradient ranging from green to red is specifically used to indicate lower-to-higher levels of genetic affinity as reported by the corresponding colour scale. c Fine-scale patterns of genetic clustering and proportions of ancestry components inferred at the individual level. The fineSTRUCTURE hierarchical clustering dendrogram obtained for Andean, Central and South American individuals included in the assembled dataset is reported, along with ancestry components inferred for each subject with ADMIXTURE analysis at K = 8. For each genetic cluster pinpointed by the fineSTRUCTURE analysis, the corresponding individuals and their admixture proportions are specifically detailed, along with the symbols used in the PCA plot for the populations they belong to. The source data used to generate the figures are reported in Supplementary Data 2—Tables 1–3.
The outgroup-f3 statistic was then computed on the assembled reference dataset to select the most suitable low-altitude control populations of South American ancestry to be contrasted with Andean groups in terms of evolved biological adaptations (see ‘Methods’ for a detailed description of the adopted selection criteria). According to this approach, while each Andean population was confirmed to share the greatest genetic similarity with the other Andean groups, Amazonian populations from Peru appeared as the next most affine ones to Andeans (Fig. 1b, Supplementary Fig. 2 and Supplementary Information). Among these putative control populations, eight Ashaninka, six Cashibo and 10 Shipibo subjects were specifically selected to constitute the low-altitude control group according to their clustering patterns and negligible Andean-specific genetic component, as pointed out by CHROMOPAINTER/fineSTRUCTURE and ADMIXTURE estimates respectively of haplotypes sharing and individual ancestry proportions (Fig. 1c and Supplementary Information).
Genomic signatures ascribable to polygenic adaptive events in Andean populations
At first, genome-wide distributions of nSL and H12 statistics, which are informative of both strong and weak selective events, were calculated for Andean and control groups relying on both WGS and imputed data. Then, we used these distributions as input for the signet algorithm to infer gene networks made up of loci belonging to the same functional pathway and that have been concurrently targeted by natural selection (see ‘Methods’). By considering only pathways confirmed by each selection statistic and by both WGS and imputed datasets, we shortlisted the most plausible biological functions having evolved adaptively in the considered populations.
Significant gene-networks identified for the low-altitude control group were found to participate primarily to immune-related pathways (e.g., Staphylococcus aureus infection, Human papillomavirus infection and Arachidonic acid metabolism), suggesting the evolution of adaptations able to mediate immune and inflammatory responses to pathogens (Supplementary Table 2). Similar selective pressures seem to have acted also on Andean groups, as attested by their putative adaptive loci belonging to the Purine metabolism and Herpes simplex virus 1 infection pathways (Fig. 2a and Supplementary Table 3).

a Circular network build with Cytoscape v3.10.3 and displaying protein-protein functional interactions inferred for the entire set of adaptive genes belonging to significant pathways identified by H12-based and nSL-based signet analyses according to both WGS and imputed datasets. Genes pointed out by LASSI-based signet analyses as participating to the Focal Adhesion/PI3K-Akt signalling pathways are also reported. Gene products that establish similar connections in the network are placed next to each other, comporting the delineation of two well-distinguishable functional clusters. In detail, non-angiogenesis related genes (e.g., belonging to the Herpes simplex virus 1 infection pathway) are reported as light-grey circles, while angiogenesis-related genes (i.e., belonging to the Focal adhesion/PI3K-Akt signalling pathways) are displayed with pink (when supported by H12 and/or nSL) and light blue (when supported by LASSI) ellipses. The source data used to generate the plot is reported in Supplementary Data 2—Table 4. b Scheme displaying functional interactions between PI3K-Akt signalling and Focal adhesion adaptive genes contributing to improved angiogenesis in Andean populations according to the KEGG database. During the early phases of angiogenesis molecular compounds made available in the extracellular matrix, such as those building the basement vascular membrane (e.g., COL4A1) and angiogenetic factors (e.g., VEGFA and PDGFD), interact with integrin and target receptors (e.g., IGF1R) stimulating protein kinases (e.g., PRKCB and PTK2), which subsequentially activate those signalling cascades essential for migration of endothelial cells and formation of new vascular structures. The yellow stars mark those genes previously identified as loci putatively mediating polygenic adaptation to hypobaric hypoxia in Tibetan/Sherpa populations52,61,62, thus providing evidence for partial genetic convergence between Andean and Himalayan adaptive traits in addition to the remarkable convergence observed at the biological function/pathway level.
Conversely, adaptive events plausibly contributing to the response to stresses imposed by the high-altitude environment were inferred only in Andeans and were mediated by genes involved in the Focal adhesion pathway, which regulates cells adhesion to extracellular matrix and subsequent signal transduction at the intracellular level (Fig. 2a, b and Supplementary Table 3). Most of the loci constituting the significant networks identified by analysing both Aymara WGS and Andean imputed datasets are indeed known to play a pivotal role in angiogenetic processes (Fig. 2a, b). Especially five of these genes (i.e., LAMA3, COL4A1, ITGB6, PRKCB and PTK2) represent the most robust candidate targets of positive selection due to patterns ascribable to polygenic adaptive evolution identified according to both nSL-based and H12-based network analyses (Fig. 2b). Among them, ITGB6, PRKCB and PTK2 have been proved to be the main regulators of vascular sprouting, which implies the transformation of blood vessels endothelial cells into cells able to migrate and proliferate out of the vessel to create new vascular structures24,25,26,27,28,29,30,31. In fact, PTK2 inactivation has been demonstrated to prevent the beginning of the angiogenetic process, leading to a lack of development of vascular tissue in embryos and thus to their premature death32,33. Although overall adaptive evolution of loci at the Focal adhesion pathway was highlighted by significant networks obtained according to both nSL and H12 statistics, the remaining 28 genes included in these networks were pointed out as potentially adaptive by a single statistic. Nevertheless, their functional relationships with the highly confirmed loci described above have been well established and they are known to contribute to the regulation of blood vessels formation as well (Fig. 2a, b). For instance, multiple studies collected evidence supporting the interaction among PTK2, VEGFA, PDGFA and PDGFD as a key driver of blood vessels formation in placenta and some variants at these loci are found to be associated with reduced functionality of maternal-foetal circulation, ischaemic placental disease and impaired foetal growth34,35.
To further validate these findings, we run the signet algorithm also on genome-wide distributions of values obtained by applying the LASSI method respectively on the WGS, imputed and low-altitude control datasets. This inferential approach was indeed proven to outperform H12 and nSL statistics in the detection of weak selective events, being also able to accurately distinguish them from hard selective sweeps36 (see ‘Methods’ for further details). After having filtered out putative adaptive gene networks shared among high-altitude and low-altitude groups, LASSI-based network analyses pointed out 39 genes plausibly targeted by positive selection and supported by results for both WGS and imputed datasets (Supplementary Fig. 3). Among them, 27 loci were found to belong to functional pathways related to angiogenesis promotion, cell proliferation and vasodilatation (Supplementary Fig. 3), with four of them (i.e., COL4A1, IGF1R, PDGFD, ITGA8) being included in the previously described significant networks belonging to the Focal adhesion pathway pointed out by H12-based and nSL-based analyses (Fig. 2b). Another significant Focal adhesion network was supported only by the Aymara WGS dataset (Fig. 2b and Supplementary Table 4), with some genes (i.e., COL4A1, COL4A4, COL9A1, LAMC2, VWF and ITGA8) participating also to a network belonging to a functionally related pathway (i.e., PI3K-Akt signalling) detected according to imputed data (Supplementary Fig. 3 and Supplementary Table 4). These results contribute to corroborate the identification of the Focal adhesion pathway and of associated molecular cascades as complex biological functions that have been shaped by the action of natural selection in Andean highlanders.
Among the adaptive genes validated by such an approach, COL4A1 encodes a type IV collagen alpha protein that is one of the main constituents of the vascular basement membrane29,37, whose degradation and remodelling represent the very first steps of angiogenesis induced by VEGF, FGF and PDGF growth factors37. Interestingly, mutant COL4A1 mice present retinal and subretinal neovascular lesions with respect to wild-type ones38, while human COL4A1 and COL4A2 variants have been associated with cerebrovascular disease29 and intracerebral haemorrhages due to systemic small-vessel disease39, suggesting that COL4A proteins play a crucial role in modulating the formation of new vascular structures. The IGF1R gene instead encodes for the insulin like growth factor 1 receptor that binds IGF1 angiogenetic factors and is known to be overexpressed in most malignant tissues where it acts as an anti-apoptotic agent by enhancing cell survival29,40. In particular, remodelling of tumour blood vessels mediated by IGF1 was observed in the melanoma mouse model40, with IGF1 and IGF1R deficiency being also associated to severe postnatal growth retardation in mice41,42,43. Similarly, homozygous partial IGF1 deletion in humans causes intrauterine growth retardation, which is accompanied by low average placental weight44. Finally, the platelet-derived growth factor PDGF-D represents another angiogenetic factor whose activity promotes tumour vasculogenesis in mice due to VEGF upregulation45. Moreover, PDGFD down-regulation was also observed in the placenta of women with preeclampsia, a syndrome characterised by high resistance utero-placental circulation, placental hypoperfusion and elevated expression of antiangiogenetic factors46.
In line with this evidence, two additional significant gene networks participating to the Apelin signalling and Vascular smooth muscle contraction pathways were proposed to have adaptively evolved in Andeans, but not in the low-altitude control group, according to both WGS and imputed data. However, these signatures did not reach statistical significance in H12/nSL-based network analyses, being detected only by LASSI-based ones and including the GNG7, PRKCE, RYR2 and RYR3 loci (Supplementary Table 4 and Supplementary Fig. 4). In particular, RYR2 and RYR3 codify for two ryanodine receptors (RyR): the former is expressed in the sarcoplasmic reticulum of the cardiac muscle29 and is essential for contraction regulation mediated by calcium release47,48, while the latter is mainly found in skeletal muscle, as well as in the heart Purkinje tissue, thus possibly contributing to electric stability of these fibres in such an organ49. When compared with acclimatised lowlanders, native Andean individuals are found to maintain heart rate unaltered during apnoea50, similarly to what observed for Himalayan people51 and suggesting that adaptive modulation of hearth frequency and cardiac output might result in a decreased risk of developing arrhythmias in these high-altitude populations.
Convergent adaptive evolution between Andean and Himalayan populations
Interestingly, when we previously applied a gene network-based approach to WGS from Tibetan and Sherpa populations, several genes included in Andean significant networks obtained from H12/nSL analyses (e.g., LAMA1, COL4A2 and ITGA2) were similarly found to participate to functional pathways targeted by natural selection (Fig. 2b, Supplementary Table 5)52. This pattern was basically confirmed by results from LASSI-based analyses. First, this approach recapitulated previous evidence supporting convergent adaptations between Andean and Tibetan highlanders involving the EPAS110 and EGLN113 genes, with genomic windows at these loci being classified as targets of selective sweeps and showing likelihood values falling in the top 1% of the distribution obtained for the Aymara WGS dataset.
As regards instead genes included in significant networks, LAMC2 was already proposed to have contributed to polygenic adaptive events also in Tibetan/Sherpa populations52 (Fig. 2b, Supplementary Table 5). This gene codifies for the laminin gamma 2 subunit, which is an epithelium-specific basement membrane protein highly expressed in different types of cancers and tumour metastasis53,54,55. Silencing LAMC2 expression was proved to cause cell cycle arrest and significantly suppresses migration and invasion of tumour cells, as well as in vivo angiogenesis of the malignant tissue due to partially blocked activation of the EGF receptor and its downstream pathway54,55. Moreover, LAMC2 expression in the placenta predominantly interests syncytiotrophoblasts and cytotrophoblasts villous and was found to be substantially upregulated in Placental Accreta Spectrum (PAS) tissues, stimulating trophoblast over-invasion via PI3K/Akt/MMP2/9 signalling pathways56. In fact, PAS is a condition causing pathologic adherence of the placenta due to abnormal trophoblast neovascularization and invasion into the uterine wall57,58. This invasion has been related to improved stimulation of pro-angiogenetic factors not only in the trophoblast embryotic tissue, but also in the maternal basal plate of the placenta58,59. We can thus hypothesise that adaptive evolution at LAMC2 and functionally related loci observed in the considered high-altitude populations might result in promoting angiogenesis in the placental tissues.
Other genes that we propose to have been targeted by natural selection in Andeans (i.e., COL4A4, PLCE1, PIK3CB and PRKCE) were previously described as plausible adaptive loci in Tibetan groups13,60,61,62 (Fig. 2b and Supplementary Table 5). Again some of them are known to be associated to inhibition of apoptosis and angiogenesis in tumours (PLCE1)63 or to cardiovascular traits (PRKCE)64. In particular, PRKCE has been demonstrated to exert a cardio-protective role against ischaemic injury64 and presents variation patterns ascribable to both adaptive evolution and archaic introgression in Tibetan populations12,60,65. Other genes instead seem to be involved in the modulation of decidua or in the decidualization phase in preparation to pregnancy66,67. For instance, COL4A4 expression, along with that of other proteins belonging to the collagen type IV family, increases during decidualization of the human endometrium67 participating to the regulation of those morphological and functional changes (e.g., increased vascular permeability, oedema, invasion of leucocytes, vascular remodelling and angiogenesis) that are essential for the establishment of a successful pregnancy68,69. Similarly, PIK3CB turned out to be up-regulated in the decidua of early-onset pre-eclampsia versus normal pregnancies, plausibly playing a role in abnormal placental development66.
According to this picture, the obtained results revealed that changes at functional pathways enabling generalised and/or placental improvement of angiogenesis, as well as cardiovascular protection, seem to have characterised the evolutionary history of both Andean and Himalayan populations51,52,65. Nevertheless, an incomplete genetic convergence between these high-altitude peoples was observed. This is not unexpected within the framework of an evolutionary scenario largely characterised by polygenic adaptive events, in which single loci play a negligible role with respect to their overall synergic interactions. Overall, we provided evidence for the same biological functions having adaptively evolved in these populations, plausibly in response to the selective pressure imposed by hypobaric hypoxia.
Putative adaptive genes associated with complex traits and eQTLs
To finally test whether the identified candidate adaptive genes are associated to complex physiological and/or pathological traits possibly implicated in the adaptive responses evolved by the considered high-altitude populations, we first consulted the Common Metabolic Disease Knowledge Portal (CMDKP) (Supplementary Table 6) (see ‘Methods’ for further details). Furthermore, we searched for expression quantitative trait loci (eQTLs) that could be able to modulate the expression of these genes in specific tissues. For this purpose, we relied on information stored in the GTEx portal and we verified if eQTLs at each gene are located in genomic windows showing large positive values of the likelihood statistic calculated by LASSI (i.e., those chromosomal regions representing the most robust targets of natural selection). The proportions of eQTLs affecting the expression of candidate genes in tissues potentially involved in the modulation of biological adaptations to high altitudes (i.e., HA-eQTLs) were reported in Supplementary Table 7, while HA-eQTLs falling in LASSI candidate adaptive windows were shortlisted in Supplementary Table 8.
According to such an approach, the most compelling results were obtained for the COL4A1, IGF1R, PDGFD, ITGA8, RYR3, LAMC2, COL4A4, PLCE1 and PRKCE genes, which turned out to be associated to haematological (e.g., haemoglobin concentration and haematocrit) and/or cardiovascular (e.g., heart rate and stroke) traits. Moreover, they included several HA-eQTLs that have been proved to influence gene expression in the aorta, heart, lung, skeletal muscle, blood and uterus (Supplementary Tables 6–8 and Fig. 3). Among them, LAMC2, PLCE1 and PRKCE showed the greatest proportion of HA-eQTLs with respect to the total number of annotated eQTLs (Supplementary Table 7). Remarkably, 100 eQTLs in the LAMC2 gene, 44 in PLCE1 and 22 in PRKCE were included in LASSI candidate adaptive windows, modulating their expression respectively in the uterus/aorta/atrial appendage, in lungs/artery tibial/atrial appendage and in the skeletal muscle and whole blood tissues (Fig. 3, Supplementary Table 8). Similarly, 51 eQTLs on the RYR3 gene, 11 on COL4A1, eight on IGF1R and four on PDGFD and ITGA8 were included in adaptive genomic windows and regulate the expression of these loci in skeletal muscle, lungs, aorta, heart and blood tissues (Fig. 3, Supplementary Table 8).
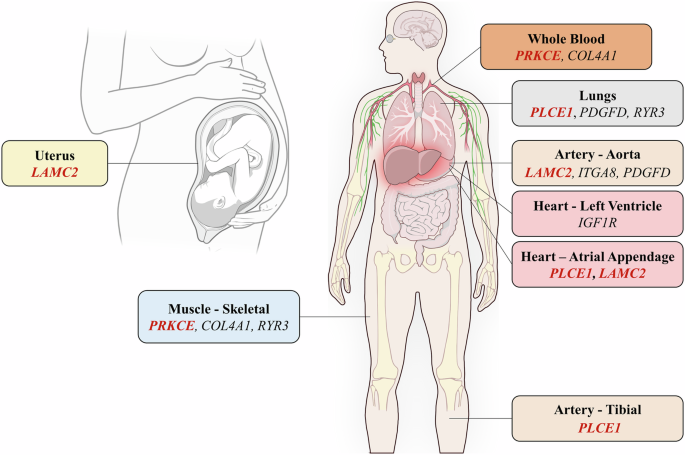
Schematic representation of the body districts in which the expression of the COL4A1, IGF1R, PDGFD, ITGA8, RYR3, LAMC2, PLCE1 and PRKCE candidate adaptive genes is modulated by eQTLs annotated in the GTEx portal. Genes reported in bold/red are those previously proposed to play an adaptive a role also in populations of Tibetan/Sherpa ancestry52,60,62,65. Illustration from NIAID NIH BIOART Source (https://bioart.niaid.nih.gov/bioart/519; https://bioart.niaid.nih.gov/bioart/420).
Overall, this evidence further reinforces the functional links observed between the inferred candidate adaptive genes and complex mechanisms that have been long proposed to be finely regulated in both Andean and Tibetan populations70,71,72. Additionally, they highlight possible associations of these adaptive changes with decreased risk of native Andean and Tibetan/Sherpa populations of developing pathological conditions that are instead more frequent in lowlanders exposed to the high-altitude stresses50,51,52,65.
Conclusions
The identified genetic signatures might contribute to the increase in the density of blood vessels that results in an improved blood flow and oxygen delivery to tissues despite the hypoxic stress, especially leading to increased uterine blood flow during pregnancy, which is an adaptive trait qualitatively (but not quantitatively) similar between Andean and Himalayan groups52,73,74. Although such a trait appears to be more enhanced in Tibetan/Sherpa populations with respect to Andean ones13,17,75, the fact that several candidate loci identified in the present study (e.g., PTK2, VEGFA, PDGFA, PDGFD, COL4A4, PIK3CB, IGF1R and LAMC2) play a pivotal role in the development of vascular tissue specifically in placenta and embryo, and thus in the establishment of efficient maternal-foetal circulation32,33,34,35,56,66,67, seems to support such an observation. Moreover, it might suggest that convergent adaptive evolution succeeded in optimising angiogenesis in these human groups mainly in early life and/or at the reproductive stage, thus leading to improved foetal development during intrauterine life (e.g., in terms of maturation of the respiratory system), which represents a key aspect to reduce neonatal mortality at high altitudes where efficiency of respiratory functions is crucial to ensure the individual’s survival. In addition, results obtained for the PRKCE, RYR2 and RYR3 genes suggest that also some cardiovascular traits might have been shaped by natural selection in both Andean and Himalayan peoples50,51. That being so, these findings highlighted polygenic mechanisms mediated by adaptive evolution of several focal adhesion and/or functionally related genes as the possible drivers of enhanced angiogenesis, oxygen transport and cardiovascular protection in Andean human groups similarly to what was previously observed in Tibetan/Sherpa populations, thus providing additional evidence for the molecular bases of their convergent adaptation to hypobaric hypoxia.
Methods
Assembled datasets
A WGS dataset including 9,439,267 SNVs for 24 Bolivian Aymara individuals who were born and live at around 3900 m of altitude was assembled from previously generated data15 and used to perform selection scans. Genome-wide genotype data were also collected for 141 individuals who were born and live between 3800 and 4200 m above sea level and belong to different Andean high-altitude populations that have been characterised by a largely shared genetic history22,23. A written informed consent for data treatment was signed by each participant as attested in previous works15,22,23 and all ethical regulations relevant to human research participants were followed. Moreover, on 09/12/2019 the Ethics Committee of the University of Bologna released approval (prot. 205142) for the present study within the framework of the project ‘Genetic adaptation and acclimatisation to high altitude as experimental models to investigate the biological mechanisms that regulate physiological responses to hypoxia’ funded by Fondazione Cassa di Risparmio in Bologna (grant n. 2019.0552). In detail, 713,014 genetic variants were retrieved for 26 Bolivian Aymaras, as well as for 21, 22 and nine subjects respectively from Aymara, Quechua and Uros populations settled in the Peruvian Titicaca Lake area23. Genotypes for 364,470 loci were collected also for 40 Quechua and 23 Aymara additional individuals22.
Data curation and imputation of the validation dataset
WGS data were submitted to QC procedures to filter for high-quality genotypes and to exclude possible closely related individuals. Specifically, the PLINK package v2.076 was used to retain only SNVs on autosomal chromosomes, variants/samples showing missing data <5% and loci with non-significant deviations from the Hardy-Weinberg equilibrium (p > 1.059 × 10−9). We also removed variants showing ambiguous A/T or G/C alleles and we calculated pairwise identity-by-descent (IBD) statistics by estimating the genome-wide proportions of shared alleles for each pair of individuals. Then, to exclude subjects related to the second degree, we filtered out one individual from each pair showing an IBD coefficient >0.270, as previously proposed for populations of Native American ancestry77 (Supplementary Information). This led to the generation of a high-quality WGS dataset including 7,966,198 SNVs and 24 individuals.
The described QC filters were applied also to the two assembled genome-wide high-altitude Andean datasets22,23 making available for validation analyses 681,932 and 364,413 variants, respectively. These data were merged by selecting only the 210,540 variants shared between both datasets and were phased by means of the Eagle algorithm v2.478 implemented in the Michigan Imputation Server (MIS) v1.2.479 using the 1000 Genomes Project Phase 3 and WGS datasets as reference panels. The MIS Minimac4 algorithm was then used to impute genotypes for 10,715,833 variants, which were submitted to the previously described QC procedures. In addition, the bcftools package v1.880 was used to retain only loci showing a squared Pearson correlation coefficient (r2) between imputed genotype probability and true genotype call >0.9581. Finally, IBD kinship coefficients were calculated for each pair of individuals in the imputed dataset using the same approach described for WGS data. According to these post-imputation QC steps, a high-density imputed dataset including 130 Andean individuals characterised for 8,024,216 variants was obtained.
Assessing samples’ genetic ancestry and consistency between non-imputed and imputed data
To verify that the collected Bolivian Aymara WGS were representative of the genetic variation observable across Andean high-altitude populations, we first explored their distribution within the overall genomic landscape of Native American populations. For this purpose, the high-quality WGS dataset was merged with a reference panel made up of genome-wide genotypes from un-admixed individuals from Central and South American indigenous populations23 and was submitted to Principal Component Analysis (PCA). Concurrently, to test also for consistency between non-imputed and imputed data, we performed PCA again by substituting the imputed data to the original ones as concerns the Andean populations included in the used Native American reference panel. In doing so, the smartpca method implemented in the EIGENSOFT package v6.0.182 was used after having filtered the dataset for variants in high linkage disequilibrium (LD). LD pruning was obtained by considering sliding windows of 50 nucleotides sites in size and advancing by five loci at the time, as well as by removing variants for each pair showing r2 > 0.2. Moreover, IBS-based matrices of individual pairwise differences in the genome-wide proportion of shared alleles were computed on both the non-imputed and imputed LD-pruned Andean datasets. These matrices were then compared by means of the Mantel correlation test83 using functions implemented in the R vegan package and empirically assessing statistical significance by running >10,000 permutations.
Selecting low-altitude control populations
To assess whether selection signatures detected in Andean people are informative about adaptations in response to high-altitude selective pressures, we selected low-altitude control populations of South American ancestry from the previously described reference panel of Native American groups23 to be used for replicating selection scans. In doing so, our rationale was to identify South American groups that: (i) share an ancient common origin with Andeans, (ii) do not show evidence of remarkable admixture with Andeans occurred after the divergence from their common ancestors, (iii) did not experience high-altitude-related selective pressures during their evolutionary history. For this purpose, we calculated outgroup-f3 statistic in the form of f3(X, Andean; CHB) to formally assess the extent of shared drift between Andean populations and the other Central and South American populations (X). This was done using the qp3Pop function implemented in the ADMIXTOOLS package v3.084 by including only groups with more than five individuals and first by considering Andean populations separately (i.e., Aymara, Bolivian Aymara, Titicaca Aymara, Titicaca Quechua, and Titicaca Uros, respectively), and then by considering them as a whole (i.e., ANDEAN group).
Moreover, to accurately select the individuals to be included in the control group, we further investigated the genetic relationships between Andeans and the putative control populations pointed out by outgroup-f3 analyses. To this end, we inferred individual-based genetic relationships by reconstructing haplotype sharing patterns by means of the CHROMOPAINTER/fineSTRUCTURE clustering approach85 and by estimating ancestry proportions for each subject using the ADMIXTURE model-based clustering algorithm86 (Supplementary Information). According to these procedures, we retained 24 individuals from low-altitude populations (i.e., eight Ashaninka, six Cashibo and 10 Shipibo), which have been characterised for 612,305 SNVs.
Inferring polygenic adaptive events from WGS and imputed data
A combination of selection scans (i.e., nSL, H12 and LASSI methods)36,87,88 suitable to detect both strong and weak selective events was applied to WGS, imputed and low-altitude control datasets. Collectively, these statistics were chosen to investigate genomic signatures left by the action of natural selection on the haplotype diversity of the examined populations, assuming that those potentially ascribable to polygenic adaptive mechanisms are represented by subtle changes in their haplotype structure and/or frequency spectrum rather than outstanding shifts in the frequency of single genetic variants. In detail, the nSL statistic was computed for each variant using selscan v.1.1.0b89 and a 20,000 bp threshold for gap scale, 200,000 bp as the maximum gap length and 4500 consecutive loci as the maximum extension parameter88. The H12 statistic was instead calculated with an ad hoc script using sliding windows of 200,000 bp and advancing by one variant at the time as previously suggested87. Unstandardised values for each statistic were then normalised across multiple frequency bins by subtracting to each of them the bin average score and by dividing the resulting value by the related standard deviation. Finally, also the LASSI likelihood statistic was computed across sliding windows, but after having estimated the average number of SNVs included in all the haplotype blocks inferred for each dataset. For this purpose, we run the –blocks function implemented in the PLINK package v2.076 separately on WGS, imputed and low-altitude control datasets. Then, the LASSI algorithm was used to calculate a distorted haplotype frequency spectrum for each window and to associate to them a likelihood statistic informative of its conformity to a model of adaptive evolution (i.e., hard/soft sweep model) or neutral evolution. Calculation of the m parameter indicating the number of sweeping haplotypes (i.e., those carrying putative adaptive variants) was used to discriminate between hard (i.e., m = 1) and soft selective sweeps (i.e., m > 1). Such a method was used to validate results from H12/nSL analyses, because it has been proved to outperform them in the identification of weak selective events according to simulations performed by considering a vast range of selection coefficients and demographic models36.
In order to test a realistic approximation of a polygenic adaptation model, we retained as the representative scores of a given gene, the highest normalised nSL and H12 values, as well as the highest LASSI score computed among those obtained for all variants/windows annotated on that gene. In doing so, the overall distribution of the LASSI statistic was filtered to select windows showing m ≥ 2 to consider only weak selective events as potential contributors to polygenic adaptations. The resulting genome-wide distributions of selection scores were then submitted to gene-network analyses using the R signet algorithm9 and by setting 20,000 iterations to generate null distributions of the highest scoring subnetwork (HSS) for each gene-network of a given size to be compared with the obtained HSS. This provided a p-value for all the identified gene networks and those <0.05 after controlling the false discovery rate (FDR) were used to point out combinations of multiple genes that participate to the same functional pathway (according to information from the KEGG database24) and that have been targeted by natural selection.
The most plausible biological functions having evolved adaptively in the Andean high-altitude groups were finally identified as those (i) supported by at least two out of the three selection scans implemented (i.e., nSL-based, H12-based and LASSI-based significant networks), (ii) replicated by applying the same pipeline of analyses to both WGS and imputed data, and (iii) not represented in significant networks obtained from the analysis of the low-altitude control dataset. Finally, to evaluate convergent evolution between Andean and Himalayan populations at a single-gene level, loci included in the supported networks were crosschecked with lists of genes whose variation patterns were previously proposed to have been shaped by the action of natural selection in Tibetan/Sherpa peoples13,52,65.
Investigating association between candidate adaptive loci and complex traits/eQTLs
The CMDKP database (https://hugeamp.org/) was used to validate associations between biological functions/traits that have been previously proposed to be involved in adaptive responses to high-altitude selective pressures51,52,65,70,71,72 and the candidate adaptive genes identified by gene-network analyses.
Furthermore, we used the GTEx portal (https://gtexportal.org/home/) to search for eQTLs at the identified candidate adaptive loci and we retained only those variants affecting their expression in tissues and/or districts of the body that have been previously proposed to be involved in the modulation of high-altitude adaptations17,90.
Statistics and reproducibility
Statistical analyses were performed using the softwares PLINK v2.0, SHAPEIT2 v2.r790, bcftools v 1.8, Eagle v2.4, MIS v1.2.4, EIGENSOFT v6.0.1, ADMIXTOOLS v3.0, CHROMOPAINTERv2, fineSTRUCTURE fs2.1, ADMIXTURE v.1.22, selscan v.1.1.0b, R v.3.6.3, LASSI GitHub repository available at https://github.com/mdegiorgio/LASSI.git. Phenotypic associations, eQTLs and pathways information in the present study were obtained consulting the CMDKP database (https://hugeamp.org/), the GTEx portal (https://gtexportal.org/home/) and the KEGG database (http://www.genome.ad.jp/kegg/). The sample sizes of each population used in the analyses are reported in Supplementary Table 1. The source data behind the graphs in the paper are listed in Supplementary Data 2.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Responses