Crystal structures of monomeric BsmI restriction endonuclease reveal coordinated sequential cleavage of two DNA strands
Introduction
Most of the bacteria and archaea possess one or several restriction/modification (R/M) systems, which consist of two distinct enzymes: a site-specific restriction endonuclease (RE) on the one hand and a corresponding methyltransferase on the other hand. The methyltransferase recognizes the same DNA sites as the coupled RE in the host’s genetic material but adds a methyl moiety to adenine (N6-methyl-adenine) or to cytosine (N4-methyl-cytosine or 5-methyl-cytosine) to protect the endogenous DNA against RE cleavage. Upon phage infection, the non-modified exogenous DNA is rapidly digested by the RE. These systems enable bacteria to discriminate between self and non-self DNA, and they can be considered as a primitive immune system1.
Among the wide variety of REs, the type II represents the most extensively studied family, with over 3500 different enzymes discovered and nearly 80 structures resolved by X-ray crystallography up to date2. These REs recognize double-stranded DNA sequences ranging from 4 to 8 base pairs in length and require only Mg(II) as a cofactor to cleave both strands either at or near their specific recognition sites3. Despite this common function, multiple sequence alignments within this family of enzymes reveal significant diversity, suggesting variations in their structures and mechanisms4. Most type II restriction endonucleases (REs) contain a PD-Xn-(D/E)XK catalytic motif that is responsible for the hydrolysis of phosphodiester bonds; however, some belong to other families, such as the H-N-H or GIY-YIG families of homing endonucleases5. Among those classified within the PD-Xn-(D/E)XK superfamily, a core structural invariant—a mixed beta-sheet flanked by two alpha-helices—can be consistently observed across all three-dimensional structures of type II restriction endonucleases available in the Protein Data Bank6. This scaffold carries the canonical catalytic motif and ensures that the important residues are maintained in the correct conformation6.
Type II RE act generally as homodimers or homotetramers and are subdivided into P, A and S subclasses based on the structure of the DNA motifs they recognize. Each monomer of type IIP RE recognizes a palindromic sequence and cleaves one of the two strands symmetrically within its recognition site. Type IIA and IIS RE can bind to non-symmetrical sequences but type IIA enzymes cleave both strands within the recognition site, whereas type IIS enzymes can cleave at least one of the two strands at a defined distance from the recognition site3. However, even inside each of these classes, the quaternary structure of REs in the absence or presence of the DNA substrate remains highly variable. Some REs are monomeric, such as AgeI7, while others are oligomeric, like BamHI and EcoRI, in the apo form8,9, but they form homodimers in the presence of DNA and cleave both strands simultaneously in a single binding event10. Others, such as MspI, BcnI or HhaI act on DNA as monomers with one catalytic site and cleave the two strands in two distinct steps11,12,13.
Here, we report the crystallographic structure of a type IIS RE, called BsmI, which is part of the R/M system of B. stearothermophilus. BsmI is notable for having two catalytic sites within a single monomer. This characteristic is shared by other enzymes, such as BtsI, BsrI, BtsCI or Mva1269I14, which are considered as fusions of ancestral heterodimers10. Like its isoschizomer Mva1269I, BsmI recognizes the sequence 5’-GAATGC-3’, cleaving the top strand one base downstream of its recognition site and the bottom strand between the G and the C of the site. BsmI features two distinct catalytic motifs: one canonical (527—PD-X17–E-X-K—548) and the other much less common (90—PD-X17–E-X12–QR—123). Interestingly, it cleaves the two strands in a single binding event but not simultaneously15,16. The presence of two catalytic sites in a single polypeptide led synthetic biologists to inactivate one of the active sites, creating the nicking endonuclease called Nb.BsmI. This enzyme can recognize the same sequence and cleave only the bottom strand17. In the following, we describe functional and structural studies of BsmI that uncover its peculiar enzymatic mechanism, which will facilitate its further rational engineering.
Results
Design of the inactive mutant N0.BsmI and structure of the complex with its cognate DNA
Based on the previous work of Zhou et al.18, we co-expressed different mutants of the BsmI RE together with the two corresponding methylases found in the genome of the B. stearothermophilus NUB36 strain in a Mrr-, McrA-, McrBC-deficient E. coli strain18. Reducing the growth temperature from 37 °C to 30 °C improved the growth rate and allowed us to achieve an appropriate yield of soluble protein after purification. A quality control of the protein samples was conducted in accordance with the ARBRE-MOBIEU guidelines19. Considering the mutations described by Xu et al. to create Nb.BsmI17 and the sequence alignment of BsmI with a few other endonucleases carrying two catalytic sites in a single polypeptide (like BtsI, BsrI, Mva1269I, BtsCI, known as class II among the type IIS REs)14,20, we designed a bottom-strand inactivated version called Nt*.BsmI and a fully inactivated version of BsmI, hereafter called N0.BsmI. For each domain, we mutated the glutamic acid of the characteristic catalytic motif PD-Xn-(D/E)-X-K (E109V, for the bottom-strand cleaving domain, and E546V, for the top-strand one).
We first obtained crystals with the fully inactivated N0.BsmI in complex with its cognate DNA in the presence of Mg2+ ions. Attempts to resolve the structure by molecular replacement were unsuccessful. The structure was solved through experimental phasing of Eu³⁺-soaked crystals, utilizing the anomalous signal from the phosphorus atoms in the DNA backbone and from the six bound europium ions, of which five were located at the external surface of the complex and one near the top-strand nicking active site. The initial electron density map had a resolution close to 2.2 Å, allowing for the construction of a complete model. The asymmetric unit contained a single monomer of BsmI and one DNA molecule tightly bound inside a central cleft, suggesting that BsmI might be monomeric in solution, similar to its isoschizomer Mva1269I15. This hypothesis was confirmed by ultracentrifugation experiments, which indicated that, in the absence of DNA, BsmI sediments as a protein of 76.8 kDa (4.6 S), while in presence of its cognate DNA, it sediments as a DNA:protein complex of 86.5 kDa (5.3 S) (Figure S1). Although traces of higher-order oligomeric states could be detected (less than 1% of the total mass), it is clear that BsmI is predominantly monomeric in solution, regardless of the presence of DNA. The theoretical sedimentation coefficients calculated from the obtained crystal structure were found to be 4.8 S in the apo-form and 5.3 S for the complex, which are close to the experimental values. These results show that the enzyme is in monomeric state both in the crystal and in solution.
Unique combination of remotely related folds for the three different subdomains
Structure analysis revealed, in accordance with previous sequence-based findings on its closely related isoschizomer Mva1269I15, that BsmI can be subdivided into three domains: (i) the N-terminal domain (in green in Fig. 1 and later) presenting a 8-strand mixed beta sheet flanked by alpha helices, (ii) a central domain (in blue) containing a smaller beta sheet, and (iii) the C-terminal domain (in orange) harboring a 10-strands mixed beta sheet flanked by alpha helices. The overall topology of the N- and C-terminal domains is similar to the invariant common structural core present in all members of the type II RE family (Figure S2). The N-terminal domain contains a catalytic motif that points toward the bottom strand, while the catalytic motif within the C-terminal domain is positioned to interact with the top strand at the level of its cleavage site. The central domain, a priori not directly involved in catalysis, will be called ”Central domain” hereafter.

A Tertiary structure of the enzyme in complex with its cognate DNA. The N-terminal contains the first mixed β-sheet flanked by α-helices and carrying the bottom-strand nicking catalytic residues. The primary and tertiary structure presents similarities with the EcoRI catalytic domain. The C-terminal contains the other mixed β-sheet flanked by α-helices and carrying the top-strand nicking catalytic residues. The primary and tertiary structure presents similarities with the FokI catalytic domain. The central domain contains a smaller mixed β-sheet surrounded by smaller α-helices. The tertiary structure presents no detectable global similarity with known structures in the PDB. B Delimitation of the three domains. The green domain corresponds to the EcoRI-like domain, also called bottom-strand nicking domain, the blue domain corresponds to the central domain and the orange domain corresponds to the FokI-like domain also called top-strand nicking domain. The dark grey chain represents the DNA bottom strand, the light grey chain the DNA top strand. C Secondary structures of the enzyme. The residue numbers delimit the β-strands and the α-helices. The catalytic motifs residues are indicated with white rounded rectangles. The grey band on the fourth β-strand of the top-strand nicking domain signals the position of the retractable “insulator loop”.
A global (whole) protein structure comparison search on the PDB using the DALI web-server21 did not find any high-confidence hits for the entire protein. However, applying the same process to the separated domains yielded interesting results for the top- and bottom-strand nicking domains. Among the proteins with a Z-score greater than 5, we identified the type II RE AgeI (PDB ID 5dwb), BsaWI (PDB ID 4zsf), NgoMIV (PDB ID 4abt) and EcoRI (PDB ID 1ckq) for the bottom-strand nicking domain; and SdaI (PDB ID 2ixs), PfoI (PDB ID 6eko), N.BsrD6I (PDB ID 5liq) for the top-strand nicking domain. Additionally, structural homologies of the top-strand nicking domain with FokI (PDB ID 1fok) and of the bottom-strand nicking domain with EcoRI (PDB ID 1ckq) are clearly visible on the views of the Fig. 2, as predicted by Armalyte et al.15.

The catalytic residues are shown in stick representation. The sequence motifs of the catalytic site are shown at the bottom. For clarity, the structural alignments were only performed between the residues [1-25, 74-142, 164-185] of BsmI (bottom strand nicking domain) and the residues [35-114, 141-170, 201-210, 235-239] of EcoRI on the one hand, and between the residues [469-499, 520-571, 597-633] of BsmI (top strand nicking domain) and the residues [417-496, 514-547] of FokI on the other hand. The RMSD was obtained with the MatchMaker tool of ChimeraX49.
Structure of the fully active wt.BsmI and the position of the divalent metal ions
Crystals of fully active BsmI, called hereafter wt.BsmI, with its cognate DNA could only be grown in the presence of Ca2+ and exhibited interesting differences compared to the N0.BsmI structure. First, the DNA is more deeply buried in the DNA-binding cleft and the top-strand nicking domain is closer to the DNA. This conformational change resulted in the loss of one symmetry axis in the crystallographic space group, transitioning from P21212 (observed in all other obtained crystals) to P21. Secondly, a notable segment (residues from 484 to 503), which includes a loop and a β-strand, is shifted so that it is threaded in the middle of the large beta sheet of the bottom-strand nicking domain. In the structure of the wt.BsmI, the loop is positioned close to the DNA minor groove, insulating the two cleavage sites from one another; in the following, we refer to it as the insulator loop. In the structures of the inactive mutants, this loop retracts to form a short beta strand, while its N-terminal part forms a loop which is mainly disordered. The sequence of insulator loop is highly conserved, as shown by the multi-alignment of the top 100 sequences obtained from a BlastP search (see below, Figure S3). Intriguingly, among the mutations introduced to create the Nb.BsmI nicking enzyme, two of them are located near the insulator loop (Gly507Asp and Arg509Val). We illustrate this conformational change with a movie (Supplementary Movie SM1), with the caveat that it should not necessarily be taken as an actual movement during the catalytic cycle, but as a dynamic illustration of the unusual type of change of conformation of this long peptide fragment.
Importantly, the wt.BsmI structure reveals the presence of three calcium ions in the two active sites. Two of these ions are located in the bottom-strand nicking active site, coordinated by Asp9, Asp91 and Glu109, while one is found in the top-strand nicking active site, coordinated by Glu470 and Asp528. These ions occupy the same positions predicted by superimposing the BamHI structure (which contains two divalent cations) onto each of the BsmI active sites (Fig. 3). Crystals of N0.BsmI could also be grown in the presence of Ca2+ ions and showed virtually no differences compared to crystals grown with Mg2+, except that the electron density more clearly showed the two divalent cations, which correspond to those seen in wt.BsmI (one in each active site).

The Nt.BsmI structure was solved at a resolution of 1.9 Å, whereas the BsmI structure was solved at a resolution of 2.85 Å. The calcium ions are represented as pink balls. The electron density was contoured at 1 sigma. A, B Details of the bottom-strand nicking catalytic domain of Nt.BsmI and BsmI crystallized in presence of calcium. Two calcium ions are present in the active site of the BsmI active enzyme whereas none are found in the active site of the inactive Nt.BsmI. C, D Details of the top-strand nicking catalytic domain. Only one calcium ion is present in both structures. E BsmI Bottom-strand nicking catalytic domain (green) structurally aligned with BamHI catalytic domain (light brown). F BsmI Top-strand nicking catalytic domain (orange) structurally aligned with the same BamHI catalytic domain. The blue spheres represent the calcium ions from BamHI pre-reactive structure (PDB id. 2BAM). The pink spheres represent the calcium ions found in wt.BsmI structure (PDB id. 9F38). The DNA represented in grey belongs to the BamHI structure.
Structures of Nt*.BsmI and Nb.BsmI mutants give an insight on the cleavage mechanism
We also solved the structure of both Nt*.BsmI and Nb.BsmI mutants in complex with their cognate double-stranded DNA substrate in the presence of calcium or magnesium ions, respectively. Nb.BsmI in the presence of Ca2+ ions did not crystallize, but the presence of Mg2+ allows to obtain crystals with DNA. However, the subtrate appears to be cleaved in the Nb.BsmI crystal. In contrast, Nt*.BsmI crystals obtained with Ca2+ and subsequently soaked in Mg2+ solutions were stable and did not show any cleavage of the DNA, consistent with an ordered reaction in which the nick in the bottom strand is a necessary step before proceeding toward the second cut. We do not observe any divalent ion in the bottom-strand nicking active sites of Nb.BsmI and Nt*.BsmI, and we see only one divalent ion (Calcium) in the top-strand nicking active site of Nt*.BsmI but none present in the Nb.BsmI site.
The structure of Nb.BsmI can be described as a snapshot of the post-reactive complex, following the cleavage of the phosphodiester bond in the bottom strand. Interestingly, despite the cut in the DNA, we do not see any conformational change compared to the inactive N0.BsmI. We observe the expected 5’-Phosphate and the 3’-OH moieties after the reaction, but the limited resolution (2.94 Å) does not provide direct insight into the presence and coordination of magnesium ions. The well-defined structure in the electron density of the Nb.BsmI:DNA complex after cleavage of the bottom strand in the crystal and its high similarity to the other structures obtained with uncleaved DNA suggests that this intermediate step in the reaction is at least partially stable and not a transient species.
In all the resolved structures of the BsmI mutants, including the Nt*.BsmI, the Nb.BsmI and the N0.BsmI presented in this article, the Insulator loop is in a retracted position, away from the DNA. Figure 4A provides a detailed comparison of the relative positions of this loop in the Nt*.BsmI and Wt.BsmI structures. In Wt.BsmI the closure of the Insulator loop brings it in contact with the DNA minor groove. A more general view is presented in Movie SM2.

A Comparison of the different positions of the Insulator loop. The wt.BsmI structure is represented in light colors, while the Nt*.BsmI one (similar to all the other mutants) is represented in darker colors. The Insulator loop is depicted in blue and its retraction movement away from the DNA is materialized by a double arrow in black. The pink spheres correspond to the calcium ions found in the wt.BsmI structure and their known glutamate ligands are in ball-and-stick representation. The DNA backbone is in grey. B Comparison of the DNA conformation with B-DNA. The Insulator loop of the wt.BsmI is represented in light blue in the context of the beta-sheet of the top-strand nicking domain (in light brown). The blue and yellow patches on the DNA backbone illustrate the position of the scissile phosphodiester bonds of the bottom and the top strand respectively. The light and dark purple DNA backbone corresponds respectively to the top and bottom strands of the cognate DNA in the ideal B-form. The Insulator loop opens up the minor groove of the DNA to bring into position the second nicking site.
DNA recognition mechanism
Comparison of the cognate DNA with standard B-DNA of the same sequence reveals that the cleavage sites are located in a slightly distorted DNA helix (Fig. 4). This distortion is characterized by an opening of the minor groove, which can be directly attributed to the closure of the Insulator loop. The two nicking domains together with the Central domain form a cleft that presents a large contact surface with DNA. Their respective interaction surface with the DNA is 1053.9, 1112.6 and 445.4 Ų. This suggests that the Central domain may have an important role in the sequence recognition specificity. As depicted in the Fig. 5A, the cleft surface facing the DNA is mainly positively charged, which is expected for a Protein:DNA binding interface. A thorough study of the refined models, including ions and water molecules, allowed us to enumerate all possible direct and water-mediated residues:DNA interactions. In Figure S4, we present a schematic diagram of these interactions, listing all the residues involved in one or more bond(s) with the DNA or with a DNA-interacting water molecule.The absence of interactions from residues in the top-strand nicking domain suggests that the complex is in a conformation compatible with bottom strand cleavage, but not with top strand cleavage. This is in line with the functional tests presented below, which indicate that cleavage of the bottom strand occurs first and is an obligatory step prior to cleavage of the top strand. An important aspect of the interactions involves the phosphate group and, occasionally the ribose moiety of the nucleotides. While these interactions do not confer specificity for the canonical recognition site, they do help stabilize the DNA interactions.
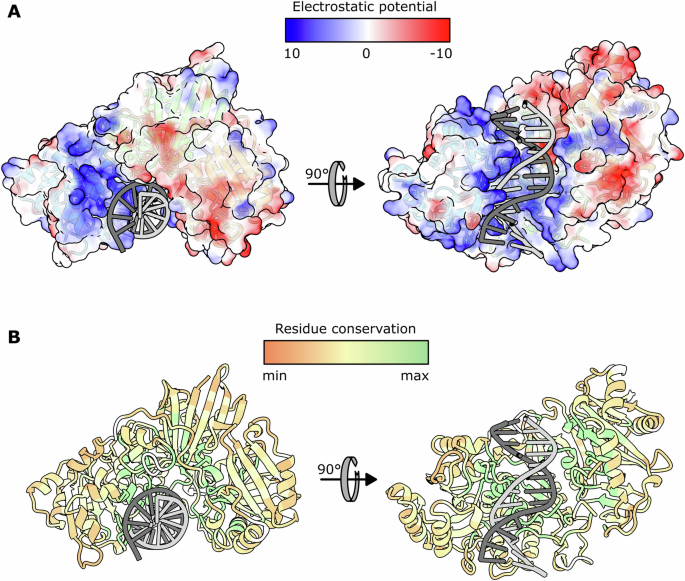
A Coulomb electrostatic potential (ESP) mapped to the molecular surface of BsmI. The color coding is shown on top, in units of kT/e. The blue surfaces have a positive electrostatic potential and the red ones have a negative electrostatic potential. On the left, the view of the same orientation as in Fig. 1B. On the right, the view of the DNA-binding cleft. The electrostatic potential was calculated with the ‘coulombic’ function of ChimeraX (v1.5)49 using default options (dielectric constant ε = 4.0 inside the protein, ε = 78.0 outside the protein temperature T = 298 K). B Residue conservation across the 100 first results of BlastP using the Consurf web server23,24. The color scale is shown on top.
All the nucleobases accommodated in the double-stranded recognition site participate in binding, sometimes redundantly, with residue side chains or backbone atoms important for the high site-specificity of BsmI. Interestingly, the first two nucleobases of the recognition site (G and A) appear to have fewer contacts than the other nucleobases, interacting only through water molecules in the Nt*.BsmI enzyme, whereas more contacts are observed in the wild-type complex. This suggests that the constraints on these two nucleobases could be more easily modified than the others, in future protein engineering experiments. Despite the important change in DNA position inside the cleft, the comparative map of Protein:DNA interactions presented on Figures S6A and S6B shows that in both conformations, the interaction is supported by the bottom-strand nicking domain and the central domain. A notable increase in the number of residues contacting the recognition site is observed in the wt.BsmI map. The loss of the water-mediated interactions can be explained by the lower resolution of the wt.BsmI electron density map, which did not allow for the placement of as many water molecules as the Nt*.BsmI structure. Non crystallographic symmetry constraints were applied during refinement, and only a few differences between the models of the two molecules in the asymmetric unit can be observed (Figures S6B and S6C).
To explain how BsmI might be impaired by methylation of the DNA, we examined close contacts between adenine (N6) or cytosine (N4) methylation sites on the DNA and the side chains of the protein. We found that the 2 central adenines (A6 on top strand and A7 on bottom strand) are recognized by the side chains of residues N118, Q119 and Q122 (Figure S4B-C), which are part of a highly conserved patch of residues (GNQAWQR in Figure S3).
Without divalent ions, Alphafold322, the last version of AlphaFold which can run predictions for the structures of complexes with DNA, ions and ligands, could not predict any complex between BsmI and its cognate DNA sequence. When divalent ions were included, the ions were correctly predicted but the DNA was positioned in the wrong orientation and shifted compared to the experimental model described here, suggesting that AlphaFold3 fails to accurately capture the essence of the interaction specificity between BsmI and its substrate (Figure S5).
Conserved motifs along the sequence and functional role of key residues
A BlastP search using the BsmI protein sequence as template allowed us to identify 184 closely related homologues with a coverage greater than 80%. These sequences were aligned using the MAFFT webserver. The multi-alignment (Figure S6) was processed with the Consurf program to highlight the pattern of conserved residues in a three-dimensional context23,24. The output is presented on Fig. 5, with the protein surface colored according to the calculated conservation levels from the multi-alignment. The multi-alignment itself reveals patches of highly conserved residues all along the BsmI reference sequence, while this representation shows how the conserved residues cluster at the surface of the cleft. All the residues involved in the DNA sequence recognition, as depicted in Figure S4, as well as those in the catalytic motifs, are highly conserved, as expected.
Figure S3 presents a Logo representation of the conserved motifs in the MSA, highlighting highly conserved regions in the alignment. We selected the eight most representative motifs and used the structure of BsmI to infer their function. Among 8 conserved motifs, two correspond to the expected catalytic motifs. While it is likely that this subfamily of monomeric RE comes from the fusion of two nucleases10, it is not known if the marginal PD-Xn-E-Xm-QR catalytic motif, which is rather unusual in the type II RE family compared to the PD-E-X-K motif, predates the fusion or not. Two other motifs correspond to an α-helix carrying residues coordinating metal ions in each active site, participating to the correct positioning of the catalytic ions inside the active site. Three motifs correspond to loops contacting either the major or the minor groove of the DNA, and are thus suspected to confer its DNA sequence specificity to the enzyme. The last region is the “Insulator loop” that appears to be mobile and to intercalate between the two catalytic sites in the active version of the enzyme. Its putative role will be commented in the discussion section.
Mva1269I, the isoschizomer of BsmI, (GenBankTM accession number AAY97906.1) ranks #181 (in term of sequence identity) among the 184 results of a BlastP run against the BsmI sequence with a coverage larger than 80% (see Table S1). The functional properties of Mva1269I have been described in ref. 15. A comparison of the two restriction enzymes sequences revealed that they share only 30.2% sequence identity, despite their functional equivalence and probable similarities in terms of mechanism (both are monomeric and carry both two catalytic sites in a single polypeptide). Alphafold2 (https://alphafold.ebi.ac.uk/) was used to generate a structural model for the protein alone with a very high confidence score all along the chain. A structural alignment by ChimeraX with BsmI revealed an overall RMSD of 3.86 Å with 644 residues (Cα aligned), and 1.275 Å for 388 pruned residues (Figure S7).
Activity tests on BsmI mutants reveal a sequential and concerted cleavage mechanism
To study the mechanism leading to the cleavage of the two DNA strands, we mutated a conserved glutamate in the top-strand nicking site (E546V – Fig. 6A), or in the bottom-strand nicking site (E109V – Fig. 6B), or in both, to independently inactivate the cleavage of each of the two DNA strands. We call these mutants Nb.BsmI (bottom strand nicking version), the Nt*.BsmI (bottom-cleavage-conditioned top strand nicking version) and the N0.BsmI (fully inactivated version).

A The top-strand nicking catalytic motif of the active version of BsmI is represented in orange while the one of the superimposed inactivated BsmI is in dark grey. The pink sphere represents a calcium ion. B The bottom-strand nicking catalytic motif of the active version of BsmI is represented in green while the one of the inactivated BsmI is superimposed in dark grey. C Design of the probe used to test enzyme activity. The strand presenting the top-strand recognition sequence is 5’-labeled with a FAM fluorophore, the bottom-strand one with a ROX fluorophore. When uncut, each strand migrates like a 59bp-long single-stranded DNA oligonucleotide. Once cut, the top strand is 30bp-long and the bottom strand is 31-bp long. D Cleavage test on a classical probe (no initial cut). The P1 control corresponds to the probe in absence of enzyme. The P2 control is a mixture of the 59bp-long bottom strand and of the 30bp-long top strand. The P3 control is a mixture of the 59bp-long top strand and of the 31bp-long bottom strand. The probe was incubated 10 min at 65 °C in presence of 15 µg/mL of N0.BsmI, Nt*.BsmI, Nb.BsmI or wt.BsmI, respectively indicated by the staggered “N0.”, “Nt.”, “Nb.” and “BsmI” annotations at the top of the lanes. The green arrow symbolizes the expected position of the cut top strand and the red one indicates the expected position of the cut bottom strand. E, F The probes presenting already a nick on the bottom or on the top strand were treated similarly to the classical one. The green and red asterisks on the left side of the bands mark the position of extra-bands due to electrophoretic artifacts. The denatured strands were migrated by electrophoresis through a 16% polyacrylamide-8M urea gel.
The activity of the different versions of BsmI was assessed using a doubly labeled fluorescent DNA probe containing the BsmI recognition site. As depicted in Fig. 6C, the top strand was 5’-FAM-labeled and the bottom strand was 5’-TAMRA-labeled. When cleaved, each labeled strand is shortened, which can be visualized by electrophoresis on a polyacrylamide gel.
As expected, wt.BsmI cleaves the two strands, Nb.BsmI cleaves only the bottom strand, and N0.BsmI leaves the two strands uncut. However, Nt*.BsmI is also completely inactive (Fig. 6D). These results support the hypothesis that the two strands are cleaved in an ordered fashion. Indeed, it appears that the bottom strand must be cut first for the enzyme to be able to cleave the top strand. To verify this hypothesis, we tested the activity of the four versions of the enzyme with probes containing a nick on either the top or bottom strand (Fig. 6E, F). Indeed, Nt*.BsmI was able to cleave the top strand of the bottom-nicked probe, consistent with the proposed two-step mechanism. Interestingly, this experiment also shows that both nicked probes can be cleaved by BsmI and that the top-nicked probe can be cleaved by Nb.BsmI.
The presence of extra bands on the gel prompted us to explore the cleavage mechanism and the migration process in more detail. These bands could result from either star activity of the enzymes or electrophoresis artifacts. Consequently, we tested the different mutants in commercial buffers where BsmI and Nb.BsmI are known to be highly specific (rCutsmart and r3.1 buffer from New England Biolabs, respectively) (Figure S8A). Since the results in the r2.1 buffer were completely similar to those obtained with the recommended buffers, we examined the migration profile of an oligonucleotide mixture mimicking the double-strand cleavage of the probe. We observed the exact same extra bands as for BsmI cleavage (Figure S8B-C), showing that the presence of the complementary strand influences the migration despite the harsh denaturating conditions in the loading dye and in the gel.
In all structures, the bottom-strand nicking domain is closer to its DNA substrate than the top-strand domain, and its active site is well positioned for the first reaction to occur. When divalent ions are visible in the electron density map (with Ca2+ ions), the catalytic residues in the bottom-strand nicking site coordinate a divalent cation that is well positioned to trigger the phosphodiester bond hydrolysis. In contrast, in the top-strand nicking site, the divalent cation appears to be too far from the DNA.
Stereospecificity for the phosphorothioate bond cleavage
In order to shed light into the cleavage mechanism, we subtituted the scissile phosphodiester bond with a phosphorothioate bond (Fig. 7A, C) in either one of its two diastereoisomers25,26,27,28. We separated the two enantiomers of a 13-base-long oligonucleotide by FPLC using a pellicular anion exchange column and a NaCl gradient (Fig. 7B). The control single-stranded DNA was eluted in a single peak, while the phosphorothioated oligonucleotide produced two distinct peaks corresponding to the RP and the SP forms of the bond. It is known that the first peak corresponds to the RP and the second to the SP enantiomer29.

A Conformation of the phophodiester bond and of the two diastereoisomers of the phosphorothioate bonds. B Chromatograms of the DNA Pac FPLC of the top strand 13-mer with phosphodiester bonds (PDE) only (in blue) and of the 13-mer presenting a phosphorothioate bond (PTO) instead of the scissile PDE bond (in green). The black curve represent the NaCl gradient applied during the enantiomers separation. The first peak is the RP and the second the SP enantiomer. C Scheme of the enantiopure probe preparation. The oligonucleotides carrying a phosphorothioate bond, purchased from an industrial manufacturer, are a mixture of the two enantiomers. They were separated by HPLC, desalted, concentrated and then ligated to other oligonucleotides as indicated on the right to form the probe. D, E The control (Ctrl) represents the probe incubated without any enzyme. The probes obtained by ligation of the top- and bottom-strand 13-mers (PDE, PTO-RP, PTO-SP) were incubated with BsmI at 15 µg/mL at 65 °C for 10 min.
The results of the test of the resistance to cleavage of the separated oligonucleotides containing a phosphorothioate bond at the scissile sites were analyzed after migration on a polyacrylamide gel. To reconstitute the same type of probes as previously, we treated the two previous strands independently by annealing and then ligating the separated oligonucleotides with a 5’-labeled strand and a downstream strand to obtain the four desired probes (top-strand RP and SP, bottom-strand RP and SP). As shown in Fig. 7D, E, BsmI cleaves the control strands and the RP enantiomer but cannot cut either the top-strand or the bottom-strand SP. Interestingly, these results confirm the proposed two-step mechanism. When the bottom-strand contains the SP phosphorothioate, both strands remain uncut. When the protection is on the top-strand, the bottom strand is cleaved while the top strand is not, consistent with the previous conclusion that the bottom strand must be cleaved before the top strand can be cut.
Normal mode analysis and the top-strand cleavage mechanism
A crystal structure does not necessarily account for the entirety of the conformations adopted by the protein along its catalytic cycle. Here, the observed conformation of the top-strand nicking active site is not compatible with a catalytic act, indicating that a conformational change is necessary for the enzyme to effectively cleave this bond. To visualize the collective degrees of freedom that allow the protein to rearrange its domains, we performed normal mode analysis using NOLB software30. The lowest-frequency mode, which is considered the most relevant at a given temperature (i.e., with the highest amplitude), clearly shows the possibility for the top-strand nicking domain to move closer to the DNA (Supplementary Movie SM2). Figure 8 illustrates the enzyme undergoing a large conformational change along the first mode, compared with the crystallographic structure. Beyond the global movement of the domain, which now aligns the top-strand scissile phosphodiester bond in the two models, the catalytic residues get closer to the DNA, suggesting that this movement could account for the top-strand cleavage mechanism. On the other hand, the normal mode analysis reveals significant DNA deformation that may be necessary to enable the movement of the protein domains. Indeed, this deformation could occur only if the bottom strand is cleaved, which explains the enzyme’s inability to cut the top strand independently of the bottom strand’s state.
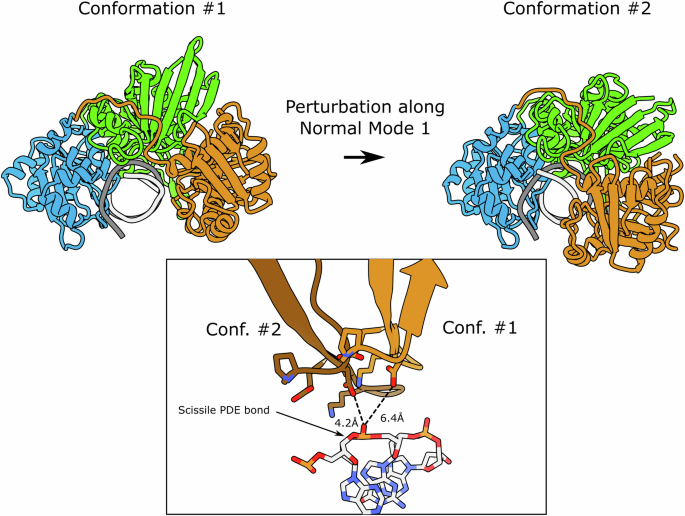
The upper panel shows the initial (#1) and final (#2) positions adopted by the domains following a deformation along the first normal mode with default value amplitude. The lower panel represents an alignment along the DNA molecules of the two models. The lighter chain corresponds to the initial conformation (the one observed in the crystal structure), the darker chain corresponds to the positions of the catalytic motif residues after deformation along the first normal mode. Catalytic residues are shown in ball-and-stick representation. The distance between the catalytic glutamate and the scissile phosphodiester bond decreases from 6.4 to 4.2 Å. Normal Modes were calculated with NOLB30.
Discussion
Here, we present the first high-resolution structure of a monomeric type IIS restriction enzyme (RE) with two active sites. To obtain the structure of wt.BsmI bound to its cognate DNA, Ca²⁺ ions were used instead of Mg²⁺, as they are catalytically inert but effectively mimic Mg²⁺ ions, at least in terms of their specific octahedral coordination geometry. This strategy of replacing Magnesium by Calcium ions has frequently been employed in structural studies of nucleic acids processing enzymes, such as DNA polymerases, for instance from the RB69 phage31. When possible, this is supplemented by structural studies of catalytically inert mutants in the presence of Mn2+ or Mg2+ 32. This approach was also adopted here, and we must be cautious in interpreting the separate effects of introduced mutations in relation to the effects of the change of divalent ions. See also33,34,35,36. Here, we can assess whether the Ca²⁺ binding sites serve as a suitable model for Mg²⁺ ions by comparing them with known structures of the same family. BsmI, as a monomeric enzyme carrying two distinct active sites, belongs to a class of RE that can be engineered to yield nicking endonuclease(s) in each of the two DNA strands, by mutating glutamate residues in their catalytic sites17,20. BsmI has actually a commercially available bottom nicking version known as Nb.BsmI, obtained by inactivating the top-strand nicking active site. Following the same principle, we demonstrated the inactivation of the bottom-strand nicking active site and produced mutants with one or both the catalytic sites inactivated. They were crystallized to structurally study the catalytic mechanism.
In the structure of wt.BsmI in presence of calcium, we observe two metal ions in the bottom-strand cleaving active site, and only one in the top-strand cleaving active site (Fig. 3C, D). The alignment of the two beta-strands carrying the catalytic residues with the ones of BamHI for the bottom-strand cleaving domain shows striking similarities in the positions of the metal ions and their ligands (Fig. 3E). In contrast, the alignment for the top-strand cleaving domain shows that one of the two sites found in BamHI is occupied by a Ca2+ ion, while the other remains empty (Fig. 3F). Remarkably, the missing metal ion corresponds to the one that is lost after cleavage in BamHI37. Since we know that the bottom strand must be cleaved to enable top-strand cleaving, we can confidently hypothesize that the cleavage of the bottom strand relaxes conformational constraints, allowing the top-strand cleaving domain-DNA complex to adopt an active conformation and to acquire the missing divalent cation. The post-reactive loss of metal ion in BamHI may indicate that the corresponding unoccupied site in BsmI becomes populated only during catalysis.
Alphafold 3 succeeded in predicting the structure of the apo-enzyme accurately and in finding the correct position of the catalytic ions in one of the 5 models generated with the default parameters but could not place correctly the cognate DNA, in fact it is modeled upside-down. It is perhaps appropriate here to alert the community of molecular biologists interested in structures or protein engineers interested in the RE specificity for DNA that the predictions of AlphaFold3 are not necessarily correct, even if its quality indicators all have a green light. It appears that crystal structures are not really dispensible at this stage, if one is interested in the atomic details of RE-DNA interactions.
Incidentally, the crystals of wt.BsmI, if optimized, could also serve as a valuable source for future detailed kinetic studies by time-resolved crystallography. Indeed, we know that the initial and ending points of the reaction can be accommodated within the same crystal packing environment. If the Ca2+ ions are carefully exchanged for Mg2+ or even Mn2+ ions, it might be possible to identify the the different steps of at least the first nicking reaction at the atomic scale.
For the recognition of the cognate DNA sequence, we find that the side chains of highly conserved residues are involved in recognition of the 3 central adenines at atomic positions where steric clashes would occur upon N6-methylation, as expected. In general, there is a very good correspondence between highly conserved motifs and functional predictions based on structural analysis (See Figure S3). BsmI, like Mva1269I, cuts the bottom strand first before cleaving the top strand, most likely in a sequential and concerted mechanism during a single-binding event. However, it is also possible to observe top-strand cleavage by Nt*.BsmI and Wt.BsmI when provided with an artificially bottom-cleaved substrate. In the Fig. 5A, we clearly observe that the central and bottom-strand nicking domains of the DNA-binding cleft exhibit a positive electrostatic potential. In contrast, part of the top-strand nicking domain in this cleft is negatively charged. We can therefore hypothesize that the DNA is initially more attracted toward the bottom-strand nicking active site. The normal mode analysis performed on the complex suggests that the enzyme is able, after the cut, to dynamically bring the top-strand-nicking active site closer to the DNA. However, this movement also requires a significant conformational change in the DNA itself. It seems reasonable to propose that nicking the bottom strand relaxes the conformational constraints in the DNA, thereby allowing a productive positioning of the top-strand scissile phosphodiester bond for cleavage by the top-strand nicking active site.
Monomeric endonucleases face a specific structural and functional challenge: they must maintain two active sites composed of three glutamates or aspartates each, located only a few angstroms apart to sequentially cleave both DNA strands. BsmI appears to have evolved to achieve cleavage in an ordered manner, with bottom-strand cleavage serving as a prerequisite for top-strand cleavage. This implies that the top-strand active site is not fully positioned with respect to DNA, and the divalent ions not fully in place, until the bottom strand is cleaved. This presents a significant electrostatic challenge, as the glutamates in the active sites are only partially counterbalanced by the divalent ions. The structure suggests that a solution may lie in an essential loop that we refer to as the “insulator loop,” which effectively isolates one active site from the other. However, this loop may also play additional and yet unknown roles, such as facilitating the translocation of the nicked DNA to the second active site. Consequently, engineering this enzyme should consider this feature and give special attention to the Insulator loop.
Rational engineering of restriction endonuclease specificity remains a challenging exercise, especially due to the high redundancy of contacts between protein residues and the DNA bases, as well as the limited structural and detailed mechanistic knowledge of the different steps leading to DNA cleavage. This high redundancy imposes a quasi-exhaustive study of epistasis for engineering the enzyme, with a potential explosion of the space to be explored that could be difficult to manage in wet experiments. Despite numerous structures of EcoRI, EcoRV, and BamHI at various stages of the cleavage mechanism, the specific transient contacts made during cleavage remain unclear.
In this study, we provide new insights on DNA cleavage by monomeric restriction enzymes carrying two active sites. Knowing the structure in several conditions, a first interesting and attainable goal could be to rationally create an active top-strand nicking enzyme that does not require prior nicking of the bottom strand. This could be explored by stabilizing the interactions between the top-strand nicking domain with DNA: (i) by relaxing some of the interactions between the bottom-strand nicking domain while maintaining the enzyme specificity or (ii) by engineering point mutations in the Insulator loop.
Methods
Protein purification
The methyltransferases operon BsmIM was codon optimized, ordered from Geneart (Thermo Fisher) and cloned in a pSB3K3 vector under the control of a Lac promoter. The plasmid was transformed into T7 Express LysY/Iq (New England Biolabs) and the cells were treated to become electrocompetent. The different mutants were obtained by site-directed mutagenesis (KLD enzyme mix, New England Biolabs) cloned into a pIVEX2.3 d vector (Roche Applied Science) and transformed in the aforementioned electrocompetent cells (Table S2). The different cultures were grown in LB at 30 °C until they reached OD 0.4–0.6 at 600 nm, they were then induced with 400 μM of IPTG and transferred at 20 °C for an overnight growth. Cell lysates were obtained using a cell disruptor (CellD) and centrifuged at 20,000 g for 30 min. The proteins purified by affinity (HisTrap, Cytiva), cation-exchange (Heparin, Cytiva) and size exclusion (Superdex 200 Increase, Cytiva) chromatography steps using the buffers described in Table S3.
Analytical Ultra-Centrifugation
Sedimentation velocity experiments were performed at 20 °C using an Optima AUC analytical ultracentrifuge (Beckman Coulter) equipped with double-UV and Rayleigh interference detection. The samples were used immediately after size-exclusion chromatography in 10 mM Tris-HCl pH 7.5, 100 mM NaCl, 5 mM MgSO4. The protein and protein-DNA complex at a concentration of 2.3 µM were spun at 42,000 rpm using an AN60-Ti rotor and 12 mm thick Epon double sector centerpieces. Absorbance and interference profiles were recorded every 4 min. Detection of concentrations as a function of radial position and time was performed by optical density measurements at wavelengths of 260 and 280 nm for proteins and protein-DNA complexes, respectively.
Buffer viscosity (η = 1.0307 cP) and density (ρ = 1.00453 g/mL) at 20 °C were estimated with Sednterp 3 (http://www.jphilo.mailway.com/download.htm). Partial specific volumes of each construct at 20 °C were estimated based on amino acid sequences using Sednterp 3. For BsmI/DNA complexes, partial specific volumes were estimated by weight averaging of the partial specific volume of the amino acid sequences (0.739 mL/g) and the nucleotide sequences (0.57 mL/g). Data were analyzed with Sedfit 16.1 using the continuous size distribution c(S) model38.
Crystallogenesis
The proteins were mixed with the 13 bp dsDNA fragment (5’-GAGGAATGCAGAC-3’) and the concentrations were adjusted to reach 128 μM (10 mg/mL) for the protein and 154 μM for the DNA. Crystallization screening trials were carried out by the vapor diffusion method using a Mosquito TM nanodispensing system (STPLabtech, Melbourn, UK) following established protocols39. Briefly, we set up crystallization sitting drops of 400 nl containing a 1:1 mixture of protein sample in complex with DNA and crystallization solutions (672 different commercially available conditions) equilibrated against 150 μl of reservoir solution in multiwell plates (Greiner Bio-One). The crystallization plates were stored at 18 °C in a RockImager (Formulatrix) automated imaging system to monitor crystal growth. Manual optimization was performed in Linbro plates with the hanging-drop method by mixing 1 µl of protein samples with 1 µl of reservoir solution. The best crystals were obtained with the crystallization condition containing 100 mM MES pH 6.5, 100 mM MgCl2 or CaCl2, 30% w/v PEG 400, 250 mM Guanidine HCl. The crystals were flash-cooled in liquid nitrogen for data collection using the crystallization solution as cryoprotectant.
X-ray data collection, structure solution and model refinement
Crystallographic data was collected at the synchrotron SOLEIL (France, Saint-Aubin) on beamlines PROXIMA-140 and processed by XDS41 with the XDSME42 or the AutoPROC43 pipeline. The first structure was solved by experimental SAD phasing using crystals soaked in a Europium Chloride solution at the Europium LIII X-ray absorption edge (1.7757 Å wavelength). A single highly redundant datasets (multiplicity of 120) was assembled by merging data collected on 5 different Eu3+ derived crystals using XSCALE41. It turned out that most of the anomalous signal was contributed by phosphorus and sulphur atoms at this wavelength. Initial substructure sites were found using the SHELX C/D/E44 pipeline and refined using Phaser45. Phases were improved by density modification using PARROT46. All the other structures were solved using molecular replacement with this model and refined with Buster47 using Coot for manual reconstruction48. Data collection and model refinement statistics are given in Table 1.
Coordinates and structure factors have been deposited in the Protein Data Bank under the accession codes (PDB id. 9EZ5) for N0.BsmI (with Mg2+); (PDB id. 9EZ7) for Nt*.BsmI (with Ca2+); (PDB id. 9EZD) for Nb.BsmI (with Mg2+); and (PDB id. 9F38) for wt.BsmI (with Ca2+). Figures showing the crystallographic models were generated with ChimeraX49.
Activity assays
The oligonucleotides were purchased from Eurofins Genomics, annealed together in ultrapure water and, when needed, assembled using the T4 DNA ligase (New England Biolabs). The reactions contained 1 μM of DNA probe, 1.5 μg/mL (20 nM) of enzyme and, except when otherwise stated, the r2.1 buffer (New England Biolabs) in which both BsmI and Nb.BsmI are known to be active. The reactions were incubated at 65 °C for 20 min, heat-inactivated at 80 °C for another 20 min before the addition of the loading solution containing 95% v/v deionized formamide and 50 mM EDTA supplemented with bromophenol blue. The products were resolved by electrophoresis on a 16% w/v polyacrylamide gel containing 8 M urea. The results were imaged using a Typhoon FLA9500 laser-scanner (Cytiva).
PTO enantiomers separation
5’-phosphorylated 13-base-long oligonucleotides containing one phosphorothioate bond were resuspended in ultrapure water. The DNA PAC PA100 pellicular anion-exchange chromatography column (Thermo Fischer Scientific) was equilibrated with a solution containing 25 mM Tris-HCl pH 6.75. After injection of 5–20 nmol of oligonucleotide, the concentration in NaCl was gradually increased from 0 to 268 mM in 2 min and then from 268 mM to 400 mM at a flowrate of 1.5 mL/min. The recovered fractions corresponding to the peaks were dialyzed against ultrapure water (ReadyLyzer 10, MWCO 1 kDa, Serva) and concentrated in a speed vacuum concentrator (Eppendorf).
Normal mode analysis
Normal Modes were calculated by the Non-Linear Rigid-Block method (NOLB) using default values for the cut-off and the amplitude, and asking for the first 10 lowest frequency normal modes30. Only the protein atoms were used in the model. 10 frames were included in the trajectory.
Statistics and reproducibility
After a broad initial screening, the crystals were optimized and reproduced twice. All the datasets presented in this article were collected on at least two different crystals for each condition. All the enzymatic activity tests were performed at least three times.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Responses