C(sp3)–heteroatom bond formation by iron-catalyzed soft couplings
Introduction
Carbon–heteroatom bonds like C–S, C–O, and C–N bonds are commonly found in natural products1,2, pharmaceuticals3,4,5, agrochemicals6,7,8, and materials9,10,11. Select examples include an acaricide named chlorbenside12, recently approved pretomanid for the treatment of multidrug-resistant tuberculosis13, and the natural product (+)-igmesine with antidepressant activity14 (Fig. 1a). The ubiquitous nature of carbon–heteroatom bonds in molecules of interest drives the discovery of methods for their general construction. Furthermore, there is an increasing demand for sp3-hybridized molecule synthesis to facilitate the discovery of functional chemicals15.
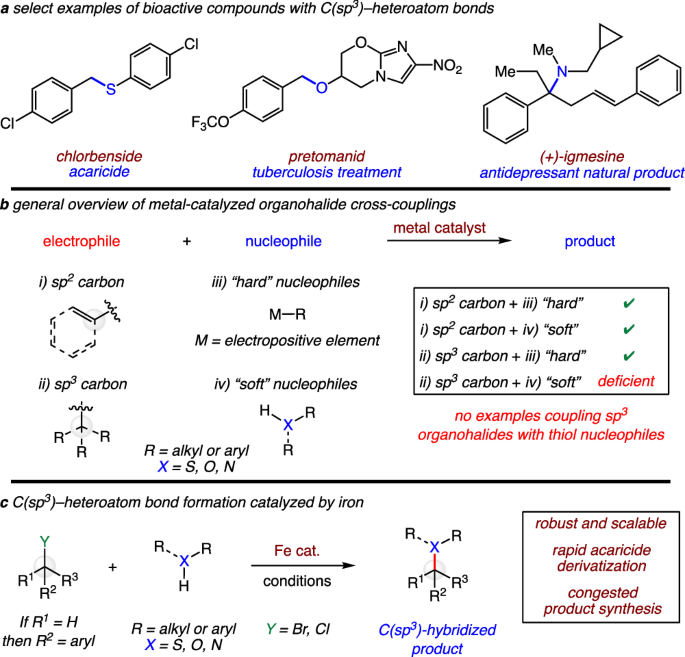
a Examples of bioactive compounds with C(sp3)-heteroatom bonds. b State of transition-metal-catalyzed cross-coupling reactions. c C(sp3)-heteroatom bond formation catalyzed by iron through cross-couplings with soft nucleophiles.
Synthetic approaches for carbon–heteroatom bond formation include nucleophilic substitution16, photochemical activation17, electrochemical activation18, and transition-metal catalysis19. Despite its extensive utility, nucleophilic substitution relies on additives that generate inorganic salts, promote undesired side reactions, or limit substrate scope. For example, despite the utility of congested (thio)ethers20 and amines21, SN2 reactions are largely limited to primary alkyl (thio)ethers because secondary alkyl halides face elimination and tertiary halides are unreactive22. As a result, congested (thio)ethers and amines are commonly synthesized using substituted nucleophiles as opposed to highly substituted electrophiles. Despite offering mild reaction conditions in comparison to nucleophilic substitution, photochemical and electrochemical approaches often face selectivity or scalability challenges23,24. It should be noted that other methods for (thio)ether synthesis using phosphine reagents, nanoparticles, and transition-metal catalysts through Lewis acid activation have been reported25,26,27,28. Although transition-metal catalyzed cross-coupling reactions between sp2-hybridized organohalides and soft nucleophiles have found broad success, couplings of sp3-hybridized alkyl halides with soft nucleophiles remain a challenge and sp3-hybridized couplings with thiols remain elusive.
Seminal studies in C–O29 and C–N30 bond construction through copper-catalyzed Ullmann-type cross-coupling reactions31 demonstrated that strategies involving transition-metal catalysts present an opportunity for carbon–heteroatom bond formation. Beyond coupling reactions with hard metalated nucleophiles for C–C bond construction32,33, several transition-metal cross-coupling reactions that produce carbon–heteroatom bonds have been developed. Reactions to form C–N34,35,36, C–O37,38, and C–S39,40,41 bonds typically engage an sp2 hybridized carbon electrophile, with sp3 hybridized carbon electrophiles facing additional β-hydride elimination complications42,43,44,45. Further, C–S bond formation is impeded by thiol oxidative S–S coupling reactions46,47, thiol-mediated catalyst poisoning48, elimination reaction pathways, and thiol-mediated C–H bond formation through radical quenching49, rendering reactions that couple sp3 organohalides with soft nucleophiles challenging to develop (Fig. 1b). Notably, the only example of a metal-catalyzed cross-coupling of C(sp3)–halides with sulfur nucleophiles is limited to benzenesulfonothioates and thiosulfonates50.
Despite nature’s ability to use iron and sulfur for target reduction51,52, iron-catalyzed cross-coupling reactions commonly couple an alkyl or aryl electrophile with a hard nucleophilic organometallic reagent. Grignard reagents for Kumada couplings in C–C bond formation have found great success53 and continue to inspire new reactivity54. In addition to reactivity, iron presents the advantage of being the most abundant transition metal in Earth’s crust55.
Given successful olefin hydrogenations in the presence of thiols56 and a cross-electrophile coupling of benzyl halides with disulfides catalyzed by iron57,58, we wondered if iron’s reactivity could be leveraged to solve the long-standing challenge of coupling C(sp3)–halide electrophiles with soft sulfur nucleophiles. This reactivity would significantly improve the atom economy in thioether synthesis and have the potential to extend into ether and substituted amine synthesis. Herein, we report the realization of general cross-coupling reactions between benzyl or tertiary halides with soft thiol, alcohol, or amine nucleophiles catalyzed by iron. The scope is broad, and the reaction is applied in large-scale synthesis, rapid herbicide analog synthesis, and in the assembly of hindered thioether, ether, and amine products (Fig. 1c). In situ reaction analysis and mechanistic studies show that the level of thiol to disulfide conversion is insufficient to account for the observed yields, highlighting that there are two distinct mechanisms when coupling an alkyl halide with a thiol or disulfide coupling partner.
Results and discussion
Hypothesis and reaction discovery
Given the lack of interaction between iron and disulfide57, we suspected that iron pentacarbonyl had the ability to activate benzylic (pseudo)halides through an oxidative cleavage event without necessitating a Grignard reagent or other hard nucleophiles. We were excited by this prospect because only a few examples of either anionic iron species or reduced iron compounds containing strong-field ligands have been reported to facilitate such reactivity to date59,60,61. This hypothesis was validated by stirring (1-bromoethyl)benzene (1) with iron pentacarbonyl to form dimer 1d in 34% yield (Fig. 2a). We then sought to promote a C(sp3) coupling with soft nucleophiles 2 without undergoing catalyst poisoning. We evaluated solvents, temperature, catalyst loading, iron sources, and additives for the coupling reaction (see Supplementary Tables 1–3). Despite observing the formation of thioether 3 when coupling (1-bromoethyl)benzene (1) with thiophenol using various iron sources (Fig. 2b, entries 2–12) and conditions, we found that iron pentacarbonyl was optimal in providing desired product (91% isolated yield, entry 12). Only a trace amount of thioether 3 is observed when running the reaction in the absence of iron (Fig. 2b, entry 1), verifying that the process is not functional in the absence of catalyst. Reducing catalyst to 5 mol% (entry 13) decreases the yield from 91 to 76%. Given the success of thioether synthesis, we were also interested in testing the coupling reaction with alcohol and amine nucleophiles 2. Despite observing only trace background reaction between benzyl alcohol and secondary benzyl halides (entry 14), it was found that 76% yield of ether formation could be achieved in the presence of catalytic iron (entry 15). Interestingly, aniline undergoes alkylation with secondary benzyl halides in the absence of iron due to the enhanced nucleophilicity of amines (entry 16), obviating the need for any catalyst in the construction of α-secondary amines (see Supplementary Table 4).

a Iron-mediated activation of organohalides. b Select results from optimization studies. a1H NMR yields were determined using 1,3,5-trimethoxybenzene as an internal standard. b10 mol% of Fe was achieved by using 5 mol% of Fe2(CO)9. cReactions were performed at 80 °C.
Scope of methodology
As shown in Fig. 3, a wide range of functional groups are applicable to the coupling between benzyl halides 4 and soft thiol and alcohol nucleophiles 5 in the presence of Fe(CO)5 to yield (thio)ether products 6. The generality across both coupling partners renders (thio)ether product 6 modular in 4 compartments. For example, unsubstituted and electron rich 4-methyl substituted thioethers 7 and 8 form smoothly with isolated yields of 91 and 85%, respectively. To emphasize the utility of the reaction, we performed a 6 mmol, gram-scale synthesis of thioether 7, which yields the desired product in 93% yield. It should be noted that overly donating groups on the arene, such as ethers, led to bromides that were unstable to chromatography. Electron-withdrawn thioethers in cyano-, nitro-, and fluoro-thioethers 9, 10, and 11 are also produced in high yield. Historically metal-reactive groups62 are unaffected and 4–, 3–, and 2-substitutions are successful, as demonstrated with fluoro-, chloro-, and bromo-adducts 11–15. Extension of the conjugated system is not detrimental to the reaction, represented by the synthesis of thioether 16 in 81% yield, and the reaction remains operable with diaryl and primary bromide substrates, demonstrated with the syntheses of 17–20 in high yields. Primary bromides also only show trace levels of background reactivity, confirming an iron-catalyzed pathway. The reaction extends beyond bromide starting materials, as exemplified by the synthesis of thioether 21 in 75% yield from the corresponding chloride starting material. A steric effect is observed when replacing the methyl group in product 7 with larger groups, as detailed with the production of thioethers 22–24. Despite the steric effect, β-tertiary thioethers 23 and 24 were isolated in 71 and 77% yield, respectively.

aReaction was performed on gram-scale. bReactions were performed at 80 °C.
The thiol coupling partner supports both aryl and alkyl thiols with varying substitutions. Thioethers bearing electron-donating and electron-withdrawing groups (e.g., 25–35), including historically metal-reactive groups or a free carboxylic acid, are isolated in high yields. Further, inclusion of nitrogen within the aromatic backbone reliably affords pyridine and pyrimidine products 36–40. Extending conjugation on the thiol group yields naphthyl thioether 41 in 76% yield. Despite observing slightly diminished conversion upon increasing the steric profile of alkyl thiols, alkyl thioethers 42–46, including a substrate with a silane group, are produced in synthetically useful yields. Functionalized alkyl thiols and electrophiles containing a fluorophore, such as a bioactive cysteine derivatives and anthracene, can be coupled to yield thioethers like 47 in 81% yield. Given the lack of an uncatalyzed reaction pathway for alcohol nucleophiles, we also developed a representative scope (48–55) for the equivalent iron-catalyzed etherification reaction. Similar modifications, including arene and alkyl variations on the benzyl halide and alcohol coupling partners, are applicable. The yields for ether production are generally lower because of notable elimination byproduct formation due to relative heteroatomic basicity; no elimination byproducts are observed in the construction of thioethers.
Synthetic applications
Interested in the successful synthesis of hindered thioethers 23 and 43, we wondered if we could leverage this reaction for the synthesis of heavily congested thioethers and ethers (Fig. 4a). To our gratification, the iron-catalyzed reaction produces sterically encumbered β-quaternary thioethers like 56 in 68% yield. Further, as represented by the production of thioethers 57 and 58 in synthetically useful yields, this method is amenable to tertiary thioether synthesis via tertiary thiol coupling. Despite being limited to benzylic substrates for primary and secondary halides, tertiary bromide coupling partners were a solution to this limitation. Using tertiary bromide as starting materials, we can also construct thioethers with tetrasubstituted carbon centers, like that found in thioether 59, in high yields. Notably, using the corresponding disulfide, analogous to our previous study57, produces congested thioether 59 in slightly lower yield and with less atom efficiency. Further highlighting the ability to form tertiary thioethers through tertiary substrate activation rather than the use of a tertiary nucleophile are the high yielding syntheses of encumbered thioethers 60–67 bearing diverse functionality. Changes in the tertiary electrophilic coupling partner also showcases versatility with the production of thioethers 68–70, including the synthesis of an ibuprofen derivative 69 in 68% yield.

a Construction of congested C(sp3)–heteroatom bonds. b Rapid synthesis of chlorbenside and analogs. aReaction was performed at 80 °C.
The coupling of an alcohol with a tertiary bromide is represented by the formation of tertiary benzyl ether 71 in 63% yield. Despite observing background reactivity between amine nucleophiles and secondary benzyl halides, we suspected that a tertiary alkyl halide that is less prone to undergo bimolecular nucleophilic substitutions would not have an uncatalyzed reaction with an amine. Control experiments (see Supplementary Table 5) confirm the lack of reactivity between a tertiary alkyl halide and an amine in the absence of catalyst, while the iron-catalyzed reaction generates sterically α-tertiary amine products 72–75 in synthetically useful yields.
Due to the vast commercial availability of various bromide and thiol coupling partners, we sought to synthesize chlorbenside (78) and analogs. When coupling corresponding aryl chlorides 76 and 77, chlorbenside (78) is isolated in 79% yield (Fig. 4b). The flexibility of the method enables the rapid synthesis of chlorbenside analogs 79–83 in high yields, emphasizing the ability to rapidly generate libraries of biologically active molecules from common precursors. While 79–81 demonstrate the facile exchange of aryl substituents, thioethers 82 and 83 introduce chemical complexity with greater steric profiles near the thioether center. It is known that some chlorbenside is excreted as the sulfoxide and sulfone equivalents63 and methylated derivatives like 82 and 83 are likely to be oxidized and metabolized at slower rates20,64.
Mechanistic studies
Mechanistic experiments of the iron-catalyzed protocol are consistent with the intermediacy of organic radicals derived from the electrophile upon activation (Fig. 5). Enantioenriched bromide 84, ent was prepared in 24 or 33% enantiomeric excess (ee) and reacted with thiophenol (85) or benzyl alcohol 86, respectively, to produce corresponding (thio)ether product 87 as a racemate (Fig. 5a). We conducted a radical spin experiment to determine if the deterioration of stereochemical information is the result of a radical intermediate (Fig. 5b). Reacting bromide 1 with thiophenol (85) or benzyl alcohol (88) in the presence of (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) under standard conditions results in a 39 or 45% (thio)ether 89 and 51 or 34% TEMPO-adduct 90 formation, respectively. An organic radical intermediate is further substantiated through the synthesis of acyclic (thio)ether 93 as opposed to cyclopropane-containing (thio)ether 92 in 76 or 100% yield when coupling radical clock substrate 91 with thiophenol (85) or benzyl alcohol (88), respectively (Fig. 5c). Each nucleophile was evaluated under their respective optimal conditions and the difference in yield is due to the difference in temperatures.

a Stereochemical examination. b Spin trap examination. c Radical clock examination. d In situ IR and thiol/disulfide interconversion studies. aReaction stirred for 36 h. b1H NMR yields were determined using 1,3,5-trimethoxybenzene as an internal standard.
Given successful iron catalysis between alkyl halides and thiols or disulfides, we used in situ infrared spectroscopy to analyze whether iron pentacarbonyl promotes the interconversion of thiol and disulfide. We were unable to track Fe–S and S–H stretches in situ because Fe–S stretches65 are found below 400 cm−1 and the S–H stretch66 was quenched in pinacolone. Fortunately, the thiophenol (85) and diphenyldisulfide (94) fingerprint region was telling. While no changes were observed with disulfide, the thiol system showed minute changes at 735 cm−1 (Fig. 5d). These observations were corroborated by NMR yields. When stirring thiophenol (85) with iron pentacarbonyl in pinacolone at 107 °C for 2 h, 5% yield of diphenyldisulfide (94) was detected. Conversely, no thiophenol (85) was detected when stirring diphenyldisulfide (94) under the same conditions. Running a reaction with 0.75 equivalents of diphenyldisulfide (94), which is the theoretical maximum loading if all thiophenol (85) were to dimerize under standard conditions, produces only 40% yield of thioether 7. Therefore, if the disulfide coupling mechanism is in action, then it does not account for the entirety of product formation and there are at least two mechanisms operating simultaneously in the thiol coupling reaction. Although specific iron species remain unknown, control experiments with 1) little reactivity when replacing iron with Lewis or Brønsted acids (see Supplementary Table 3), and 2) no Markovnikov 1,2-addition when using styrene in place of bromide 1 (see Supplementary Fig. 39) are consistent with iron directly activating the substrate through an oxidative cleavage event.
In conclusion, we have extended metal-catalyzed cross-coupling reactions to the coupling of C(sp3) benzyl or tertiary alkyl halides with soft nucleophiles. The reaction is catalyzed by iron and avoids the use of exogenous acid or base. Good efficiency with a broad steric and electronic generality for each coupling partner is observed. The system can be used for C–S, C–O, and C–N bond construction, gram-scale synthesis, and highly congested C(sp3)–heteroatom bond formation. The use of hindered tertiary alkyl halides breaks through the benzylic substrate limitation, and the reaction’s generality enables rapid synthesis of compound libraries, exemplified by the synthesis of chlorbenside and its analogs. Results from mechanistic experiments are consistent with a stereoablative pathway that likely involves an organic radical intermediate. In situ analysis and interconversion studies also point to two distinct mechanisms when coupling benzyl or tertiary alkyl halides with either a thiol or disulfide coupling partner. Due to the importance of C–S, C–O, and C–N bonds in various fields and significant interest in developing metal-catalyzed reactions, we expect this advance to be of interest to the broader scientific community.
Methods
General considerations
Unless stated otherwise, reactions were conducted in flame-dried glassware under an atmosphere of nitrogen using anhydrous solvents (freshly distilled or passed through activated alumina columns). All commercially obtained reagents were used as received unless otherwise specified. All work-up and purification procedures used reagent grade solvents purchased from Fisher, VWR or Sigma-Aldrich. Reagents and catalyst used were purchased from the following vendors and used as received: diphenyl disulfide (99.9%), iron (III) acetylacetonate (99.9%), triphenyl phosphine (99%), carbon tetrabromide (99%), bis(cyclooctadiene)nickel(0) (95%), phenylethanol (98%), pinacolone (98%), magnesium chloride (98%), aluminum chloride (99%), benzyl bromide (98%), 4-chlorobenzaldehyde (97%), tetrakis(triphenylphosphine)nickel(0) (95%), phenylacetic acid (99%), aniline (99.5%), N-methylaniline (99%), thiophenol (97%), benzaldehyde (99%), pivaldehyde (96%), phenylmagnesium chloride (2 M in THF), ethylmagnesium chloride (3 M in ether), triethylamine (99.5%), cyclohexylmagnesium chloride (1 M in MeTHF), methylmagnesium chloride (3 M in THF), methylmagnesium iodide, lithium bromide (99%), hydrogen bromide (48% in water), nitropropane (98.5%), ethyl acetoacetate (99%), ethyl malonate (99%), naphthalen-2-yl-ethanol (98%), 2-methyl-1-phenylpropanone (97%), tetrakis(triphenylphosphine)palladium(0) (99%), trimethylsilyl bromide (98%), and trimethylsilyl chloride (98%) were purchased from Sigma Aldrich. Iron (II) chloride (99.5%), iron(III) bromide (98%), iron (0) pentacarbonyl (99.5%), chlorobenzenethiol (97%), 4-dimethylaminopyridine (99%), benzhydryl bromide (90%), and copper (II) bromide (99%) were purchased from Alfa Aesar. Bromoethylbenzene (95%), 4-cholorobenzenethiol (98%), 4-nitrobenzenethiol (95%), ethyl mercaptan (98%), cyclohexanethiol (98%), 4-mercaptobenzonitrile (98%), 4-mercaptobenzoic acid (95%), 4-chlorobenzylmercaptan (98%), 2-propanethiol (96%), 2-bromobenzaldehyde (98%), 3-bromobenzaldehyde (98%), 4-bromophenylethanol (97%), 4-acetylbenzonitrile (97%) were purchased from TCI chemicals. N,N’-diisopropylcarbodiimide (>99%) was acquired from Advanced ChemTech. Anthracenyl bromide (98%), 1-bromoethyl-4-fluorobenzene, iron(II) trifluoromethanesulfonate (98%), iron(III) trifluoromethanesulfonate (98%), Ibuprofen (99%), 5-trifluoromethyl-2-mercaptopyridine (98.87%), 3-bromo-6-mercaptopyridine (95%), 4-methoxythiophenol (97%), 3-methoxythiophenol (97%), 2-methoxythiophenol (95%), 2-pyridinethiol (97%), 2-naphthalenethiol (98%) and (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (97%) were obtained from Ambeed. Iron (III) chloride (98%), iron (II) bromide (97%), diiron nanocarbonyl (98%), 2-methyl-2-propanethiol (99%), toluenethiol (98%), 4-fluorothiophenol were purchased from Thermo Scientific. Iron (II) fluoride (99%), iron (III) fluoride (99%), and 4-benzenethiol (98%) were purchased from Strem Chemicals, and 4-(trifluoromethyl)thiophenol (95%) were purchased from Matrix Scientific. Reaction temperatures were controlled using IKA Plates (RCT digital) and the built-in temperature modulators. Thin layer chromatography (TLC) was conducted with EMD gel 60 F254 pre-coated plates (0.25 mm) and visualized using a combination of UV light, potassium permanganate, phosphomolybdic acid, and p-anisaldehyde staining. Silicycle Silica flash P60 (particle size 0.040–0.063 mm) was used for flash column chromatography. 1H NMR spectra were recorded on a Mercury (400 MHz), or Varian spectrometers (500, 600 MHz) and are reported relative to deuterated solvent signals. Data for 1H NMR spectra are reported as follows: chemical shift (δ ppm), multiplicity, coupling constant (Hz) and integration. 13C NMR spectra were recorded on Mercury (100 MHz), or Varian spectrometers (125 MHz, 150 MHz) and are reported relative to deuterated solvent signals. 19F spectra were recorded on a Varian spectrometer (564 MHz). IR data were collected on an Agilent Cary 630 FTIR Spectrometer and Mettler Toledo ReactIR 720 L. All IR data are reported in terms of frequency absorption (cm–1). Melting points were recorded on a VWR melting point apparatus, high resolution mass (HRMS) spectra were obtained on an Agilent 6545Q-TOF LC/MS, and chiral SFC data were collected on an Agilent 1260 Hybrid SFC/UHPLC system equipped with the following chiral columns: ChiralPak IA-3, 4.6 × 250 mm, 3 mic; ChiralPak IB N-3, 4.6 × 250 mm, 3 mic; ChiralPak IC-3, 4.6 × 250 mm, 3 mic; ChiralPak IH-3, 4.6 × 250 mm, 3 mic; ChiralPak IG-3, 4.6x250mm, 3 mic; ChiralPak IJ-3, 4.6 × 250 mm, 3 mic; and ChiralPak IK-3, 4.6 × 250 mm, 3 mic.
Representative procedure for the synthesis of a bromide substrate
To a flame-dried 50 mL round-bottom-flask equipped with a stir bar was added 2-bromobenzaldehyde (926 mg, 5.0 mmol, 1.0 equiv.) and THF (4.0 mL). The mixture was cooled to 0 °C under a positive pressure of N2 and MeMgCl (3.0 M in THF, 3.3 mL, 10.0 mmol, 2.0 equiv.) was then added dropwise over 5 min. The mixture was stirred for additional 20 min at 0 °C. The reaction was quenched with saturated aqueous NH4Cl (10.0 mL) and extracted with EtOAc (10.0 mL x 3). The organic layers were combined, dried over Na2SO4, and concentrated under reduced pressure. The resultant oil was purified by silica flash chromatography (1:4 EtOAc:hexane) to yield the corresponding alcohol as a pale-yellow oil (667 mg, 66% yield).
The resultant alcohol was added to a flame-dried 50 mL round-bottom-flask equipped with a stir bar. CH2Cl2 (10.0 mL) was added, and the solution was cooled to 0 °C. PBr3 (156 μL, 1.7 mmol, 0.5 equiv.) was added to the solution dropwise, and reaction mixture was further stirred for 30 min at 0 °C. The reaction was quenched with water (10.0 mL) and extracted with CH2Cl2 (10.0 mL × 3). The organic layers were combined, dried over Na2SO4, and concentrated under reduced pressure. The resultant oil was purified by silica flash chromatography with 100%hexane to yield the desired bromide as a light-yellow oil (573 mg, 65% yield).
Representative procedure for the iron-catalyzed thiol (or amine) coupling
To a flame-dried 4 mL dram vial equipped with a stir bar was added Fe(CO)5 (10.0 mg, 0.025 mmol, 6.8 uL, 10 mol%) in a glove box. The vial was taken out of the glove box and placed under N2 in a fume-hood. To the vial was added pinacolone (1.0 mL), bromide coupling partner (0.5 mmol, 1.0 equiv.), and thiol (or amine) (0.75 mmol, 1.5 equiv.). The reaction vessel was sealed with a teflon screw cap and the mixture was stirred at 107 °C for 24 h. The mixture was quenched with water (1.0 mL) and the aqueous layer was extracted with diethyl ether (3 × 1.0 mL). The organic layers were combined, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The resultant mixture was purified by silica flash chromatography to yield the desired thioethers.
Representative procedure for the iron-catalyzed alcohol coupling
To a flame-dried 4 mL dram vial equipped with a stir bar was added Fe(CO)5 (10.0 mg, 0.025 mmol, 6.8 uL, 10 mol%) in a glove box. The vial was taken out of the glove box and placed under N2 in a fume-hood. To the vial was added pinacolone (1.0 mL), bromide coupling partner (0.5 mmol, 1.0 equiv.), and alcohol (1.5 mmol, 3.0 equiv.). The reaction vessel was sealed with a teflon screw cap and the mixture was stirred at 80 °C for 24 h. The mixture was quenched with water (1.0 mL) and the aqueous layer was extracted with diethyl ether (3 × 1.0 mL). The organic layers were combined, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The resultant mixture was purified by silica flash chromatography to yield the desired ether.

Responses