Diabetic foot exacerbates gut mycobiome dysbiosis in adult patients with type 2 diabetes mellitus: revealing diagnostic markers

Introduction
Over the last two decades, diabetes mellitus (DM) has been a growing global public health threat [1, 2]. Type 2 diabetes (T2DM) is a condition where there is a disruption in insulin production and/or resistance to insulin in the body, along with factors like obesity and genetic issues. This results in problems with how the liver, muscles, and fat tissues use glucose [3]. According to the most recent information from the International Diabetes Federation (IDF), there were 537 million individuals between the ages of 20 and 79 with diabetes in 2023, with 90% of them having type 2 diabetes. This number is projected to rise to 783 million by 2045. In China, on the one hand, this is due to irrational eating habits, such as a refined diet, with grains based on white rice and noodles containing little fiber, and a meat-based diet with fewer vegetables and fruits. On the other hand, it is sedentary and lack of exercise. It is coupled with genetic factors and the increasing trend of population aging. Result in 140.9 million people aged 20–79 with DM in China, with an incidence of 12.8%, accounting for 26.26% of the global number of patients with DM. Part of the population does not pay enough attention to DM, not only in pre-disease testing but also in post-disease treatment as well as aftercare. It is expected that by 2045, China’s patients with DM will reach 174.4 million, and it has become the first primary chronic disease in China.
Diabetic foot (DF) is a severe long-term complication of DM [4], primarily due to vascular issues, nerve damage, infections, and metabolic issues, leading to poor blood flow to the foot and tingling sensations [5]. Minor injuries may have prolonged healing times due to insufficient oxygen and nutrients. It may severely affect muscles and bones, eventually leading to tissue necrosis or amputation [6]. This disease has a high rate of amputation and mortality and is long-lasting and difficult to recover from [7]. It is estimated that every 20 s, a patient with DM suffers an amputation due to DF worldwide, with a mortality rate as high as 22%. Therefore, prevention is the primary treatment for DF, and an improved success rate of early screening can standardize follow-up treatment and significantly improve prognosis.
Microbiome research has focused predominantly on gut bacteria. However, the gut microbiota also contains commensal fungi, collectively known as mycobiome. Researchers have limited knowledge about the fungal populations in the gut, which play a significant role. The mycobiota were identified as part of the typical gut flora in 1967 [8]. Approximately 70% of healthy adults have fungi in their gastrointestinal tract, usually at a concentration of up to 1000 fungal cells per milliliter or gram of intestinal contents [9]. However, fungal populations comprise less than 1% of the total gut microbiota. Changes in the fecal fungal microbiome have been described in metabolic and endocrine-related diseases such as alcohol-associated liver disease [10] and non-alcoholic fatty liver disease [11]. In 2021, Doron et al. [12] found that commensal fungi in the intestines of mice and humans induced the production of antifungal antibodies in the blood and that CARD9, an important regulator of the intrinsic immune system, played a crucial role in the production of antifungal antibodies, which could protect the host from, and attenuate, lethal fungal infections. Other studies have also reported intestinal fungal profiles in middle-aged and elderly populations, analyzing the differences in fungal composition and their association with aging in middle-aged and elderly populations [13]. In a recent study, it has been found that the more complex the network of intestinal fungal interactions, the more severe the deterioration of glucose metabolism. Fusarium may be a key culprit in the alteration of the intestinal fungal network, which interferes with the insulin signaling pathway by affecting the levels of various metabolites, exacerbating insulin resistance [14]. A recent study published in CELL has established the largest reference genome catalog of human cultured intestinal fungi to date through large-scale culture sequencing of human intestinal fungi, completed metabolic function analysis, revealed the diversity of human intestinal fungi and their potential roles in common diseases, and expanded the existing intestinal fungal genome resources, which provides important reference data for the study of the intestinal fungal community structure, biological functions, and other aspects. It provides important reference data for the study of the structure and biological function of intestinal fungal communities [15]. These studies show that fungi play a crucial role in the gut ecosystem and may significantly affect the host’s health as a vital part of the microbial community [16]. Various fungal populations have been discovered throughout the human gastrointestinal tract, with a predominant presence of Ascomycota, Basidiomycota, Zygomycota, and Chytridiomycota [17,18,19]. Candida, Saccharomyces cerevisiae, and Malassezia are the most common fungal species in the human gut [20]. Due to the influence of living environment and dietary habits on the composition of intestinal fungi, no study has yet reported the differences in the composition of intestinal fungi among patients with DM in Yunnan, China, especially those who have combined DF. Hence, the composition of fungi and their potential role in patients with T2DM and T2DM-DF in Yunnan need to be further understood.
Therefore, here, we performed fungal microbiome analysis of fecal samples from 33 patients with T2DM and 32 patients with T2DM-DF from local, as well as 32 non-obese subjects without any known disease as healthy controls (HC), to investigate the fungal microbiome composition in HC, T2DM, and T2DM-DF. The differences between fungal relative abundance in diseases and possible functions were evaluated to gain a deeper insight into variability in fungal composition. And will help to enrich the mapping of the differences in gut fungal composition in patients with DM and DF and provide the possibility of precise diagnosis and treatment for these patients.
Materials and methods
Patient inclusion and ethical statement
The study was conducted after obtaining the necessary ethical approval from Ethics Research Committee of Affiliated Hospital of Yunnan University.
All the eligible patients had an established diagnosis of T2DM and T2DM-DF, and the subjects included men and women. The T2DM inclusion criteria were the following: (1) age 20–75 years; (2) meeting the 1999 World Health Organization (WHO) diagnostic criteria for T2DM; (3) HbA1c (NGSP) ≥ 6.0 but <9.0%; (4) BMI ≥ 25.0 kg/m2; and (5) diet and exercise or medication only. The T2DM-DF inclusion criteria were the following: (1) age 20–75 years; (2) the diagnosis of DF was in accordance with the criteria of the Chinese Guidelines for the Diagnosis and Treatment of Diabetic Foot, and Wagner grading was performed. The T2DM exclusion criteria were (1) having type 1 diabetes and (2) not taking diet pills or hypoglycaemic drugs (except metformin). The T2DM-DF exclusion criteria were (1) tissue defects or lesions (ulcers or gangrene) below the ankle in non-diabetic patients and (2) not taking diet pills or hypoglycaemic drugs (except metformin). The both T2DM and T2DM-DF exclusion criteria were (1) systemic antibiotics or a laxative within 6 weeks before inclusion; (2) use of probiotics, prebiotics, and synbiotics within 3 months before inclusion; (3) failure to take supplements as prescribed by a doctor; (4) currently taking non-steroidal anti-inflammatory drugs, antipsychotics or niacin; (5) daily alcohol consumption >30 g; (6) suspected or definite history of substance abuse; (7) current smokers; (8) food allergy; (9) those who develop acute complications such as diabetic ketoacidosis or hyperosmolar coma or severe recurrent hypoglycemia; (10) gastrointestinal disorders such as food allergies, coeliac disease, irritable bowel disease; (11) with gastrointestinal surgery; (12) significant immunodeficiency; (13) serious kidney disease serious; (14) liver disease excluding fatty liver; (15) known cardiac valvular disease; (16) diseases of the nervous system; (17) malignant tumor; and (18) breastfeeding or pregnancy. In addition, considering the influence of daily diet on gut fungi, we ensured participants had relatively similar diet structures by questionnaires. Namely, nutritional habits will be assessed using a standardized 14-day recall questionnaire, which will be discussed with a dietician.
Based on this, 33 local subjects diagnosed with T2DM and 32 local subjects diagnosed with T2DM-DF were chosen from the endocrinology clinic. Additionally, we found 32 non-obese subjects without any known disease, otherwise healthy local individuals as healthy controls.
The research was conducted following appropriate guidelines and regulations set by the committee. All volunteers received an information sheet explaining the study’s purpose, design, and confidentiality. Written informed consents were acquired.
We collected basic information from all volunteers and took their blood samples. We also provided all subjects with a sterile fecal specimen container with instructions and a collection spoon. A total of 97 blood samples were collected for biochemical parameters testing. Ninety-seven fecal samples containing 2 to 4 grams of recently passed feces were gathered in sterile receptacles. Specimens were promptly preserved in liquid nitrogen and then moved to a temperature of −80 °C for storage until additional testing. Participants with watery (diarrheal) feces and recent antibiotic usage in the past 3 months were not eligible for inclusion in this research.
Biochemical analysis
We tested a total of 20 biochemical parameters, including alanine aminotransferase (ALT), aspartate aminotransferase (AST); total bilirubin (TBIL); direct bilirubin (DBIL); unconjugated bilirubin (UBIL); total cholesterol (TC); Triglyceride (TG); high-density lipoprotein cholesterol (HDL-C); low-density lipoprotein cholesterol (LDL-C); fasting blood glucose (FBG); glycated hemoglobin (HbA1c); blood urea nitrogen (BUN); white blood cell (WBC); neutrophil granulocyte percentage (N%); lymphocyte percentage (L%); absolute neutrophil count (ANC); lymphocyte (LYM); red blood cell (RBC); hemoglobin (Hb); blood platelet (PLT). All of which were completed by the clinical laboratory.
DNA extraction
Following the manufacturer’s instructions, DNA was extracted from feces using the QIAamp DNA Stool Mini Kit (Qiagen, USA). To ensure successful DNA isolation, the amount of DNA was measured using a NanoDrop ND2000 spectrophotometer. The quality of the sample was then determined by agarose gel electrophoresis. Sterile water was also used as a negative control. The extracted DNA was stored at −80 °C for further analysis.
Illumina 18s rRNA amplicon sequencing
Polymerase chain reaction (PCR) was used to amplify fungal 18s rRNA genes. Primers containing a unique barcode were custom-made to match the designated sequencing area. PCR was conducted with TransGen AP221-02 and TransStart Fastpfu DNA Polymerase (ABI GeneAmp® Type 9700). The PCR samples were then measured with the QuantiFluor™-ST Blue fluorescence quantification system (Promega, USA) based on the initial electrophoresis results and combined with the sequencing volume needed for each sample. Finally, Illumina library construction and sequencing were completed.
Bioinformatic analysis
QIIME and the R package version 3.5.1 were employed to assess fungi, with the Kruskal–Wallis test utilized for initial comparisons, followed by pairwise Mann–Whitney U tests to analyze differences in alpha diversity indices and fungal abundance data across distinct groups. Subsequently, the p values were adjusted using the Bonferroni correction method. Furthermore, a Venn diagram was constructed to illustrate the common intestinal fungal species present in 78% of the samples from each group, and Beta diversity analysis was conducted to examine the structural differences in fungal communities among samples utilizing UniFrac distance metrics, which were then visualized through principal component analysis (PCA).
Furthermore, variations in the UniFrac distances between groups were assessed through analysis similarities (ANOSIM). IBM SPSS Statistics software 19.0 was used to conduct a one-way analysis of variance (ANOVA) and post hoc most minor significance tests to compare the abundance of taxa at different levels (phylum, family, genus, and species) within the groups. Additionally, LEfSe, which stands for linear discriminant analysis (LDA) effect size, was utilized to discover biomarkers for prevalent taxa and functional pathways by determining the LDA score (greater than 4) among different groups. The heatmap was generated with the R package version 3.5.1. Additionally, Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt), a tool that reconstructs unobserved states in communities based on OTUs, was used to predict enzyme functionality using the Kyoto Encyclopedia of Genes (KEGG). BugBase was also utilized to predict fungal composition from the sequencing data. Data are presented as mean ± standard error of the mean (SEM) with a significance threshold of p < 0.05.
Statistical analysis
Except for the 18s rRNA data described in detail, the data obtained in this study are presented as the mean ± SEM. The statistical analysis was also conducted using IBM SPSS Statistics software version 19.0 on a Windows platform, with distinctions between the three groups evaluated through the independent-sample t-test. Furthermore, a one-way analysis of variance (ANOVA) was conducted to assess multiple groups, with subsequent Newman–Keuls post hoc tests performed. It is essential to mention that a p value of 0.05 was deemed statistically significant.
Results
The clinical laboratory parameters in T2DM and T2DM-DF
We recruited 33 individuals with T2DM (average age 46, 57.5% male), 32 individuals with T2DM-DF (average age 54, 75% male), and 32 non-obese subjects without any known disease as an HC (mean age 43 years, 27% male). All participants submitted blood and fecal samples. As expected, patients with T2DM had a significantly higher body mass index (BMI) than HC. Conversely, those with T2DM-DF did not show a clear distinction in BMI compared to HC but did have a significantly lower BMI than individuals with T2DM. It is well known that a higher BMI represents a higher obesity index. In contrast, patients with T2DM-DF have a lower BMI than T2DM, which may be due to disorders of glucose metabolism, insulin resistance, prolonged dietary control, or medication side effects resulting from a prolonged diabetes process.
Some clinical laboratory parameters were significantly altered in T2DM and T2DM-DF (Table 1). Among these, AST and BUN were only markedly higher in patients with T2DM-DF than in HC. Elevated AST indicates liver damage, while BUN indicates kidney function. A proportion of patients with T2DM-DF are associated with liver and kidney damage, which may be caused by diabetic neuropathy, diabetic vasculopathy, diabetic nephropathy, and co-infections. TBIL and TG were significantly lower in T2DM-DF than in T2DM. TBIL is also a marker to evaluate liver function. The decrease in TG is due to inadequate lipid intake in patients with T2DM-DF due to a more prolonged unbalanced diet or due to excessive use of hypoglycaemic drugs. And UBIL, TC, LDL-C, RBC, and Hb were noticeably expressed only in T2DM-DF (Table 1).
This suggests that T2DM and T2DM-DF showed significant differences in relevant clinical laboratory parameters.
The effect of diversity and richness of gut fungal community between HC, T2DM and T2DM-DF
To determine whether the disease severity or other clinical metadata of T2DM and T2DM-DF are related to gut fungi, total DNA samples were extracted from fecal specimens obtained from all groups, followed by Illumina 18s rRNA amplicon sequencing and fungal taxonomic classification. All samples were correctly sequenced. After filtering the qualified reads, all samples’ quality-filtered reads were 7,026,239, and the average amplicon length was 241 bp. The raw sequencing data are available on request. The results showed that the sequencing reads were enough to represent most of the fungal community.
Next, differences in the richness and diversity of fungal communities among the three groups were assessed using α-diversity. The α-diversity was presented as four indexes: Chao index and ACE index estimate the community richness; Simpson index and Shannon index estimate the community diversity. Compared with the HC, T2DM and T2DM-DF showed a significant decrease in both the Chao index and ACE index, but the reduction in T2DM-DF was more prominent. Simpson index and Shannon index of T2DM were markedly changed compared with T2DM-DF, while there was an inapparent from HC. However, T2DM-DF was notably different from the HC (Fig. 1a).

The T2DM and T2DM_DF gut fungal changes on (a) α-diversity were determined using Chao index, Ace index, Simpson index and Shannon index; b Venn diagram illustrated overlap of OTUs in gut fungi among the samples; c, d β-diversity was determined using Euclidean distance based principal component analysis (PCA) and weighted UniFrac distance based principal coordinates analysis (PCoA). Data are expressed as means ± SEM. *p < 0.05; **p < 0.01 and ***p < 0.001.
A Venn diagram overlaps provided a better understanding of the shared richness among groups. Only 135 out of the total 1596 OTUs were found in all groups, with the remaining OTUs distributed among three groups, two groups, or within each group (Fig. 1b). Moreover, OTUs in T2DM and T2DM-DF were significantly reduced compared with HC (Fig. 1b).
The β-diversity reflects the difference in fungal community composition among the three groups. A principal component analysis (PCA) (Fig. 1c) plot and principal coordinates analysis (PCoA) (Fig. 1d) were conducted. The variance analysis showed a significant difference in the overall fungal community composition among various groups.
These data indicate that the T2DM and T2DM-DF remarkably reduced the richness and diversity of fungal communities, and it was also different in T2DM-DF compared to T2DM.
Compositional changes of the gut fungal community in patients with T2DM and T2DM-DF
The gut fungal profiling from HC, T2DM, and T2DM-DF revealed two significantly altered phyla, Ascomycota and Basidiomycota, of which two accounted for 99% of the fungal community (Fig. 2a). In the HC, Ascomycota was the dominant phylum, followed by Basidiomycota (Fig. 2a). The abundance of Ascomycota in T2DM-DF was significantly higher than that in HC and T2DM (Fig. 2b). In addition, the abundance of Basidiomycota in T2DM-DF was considerably lower than in HC and T2DM (Fig. 2c). Interestingly, the Ascomycota/Basidiomycota ratio was notably altered in both T2DM and T2DM-DF compared with HC, and a significant increase in the Ascomycota/Basidiomycota ratio occurred in T2DM-DF compared with T2DM (Fig. 2d). That is to say, the gut fungi in T2DM-DF were changed more drastically at the phylum level.
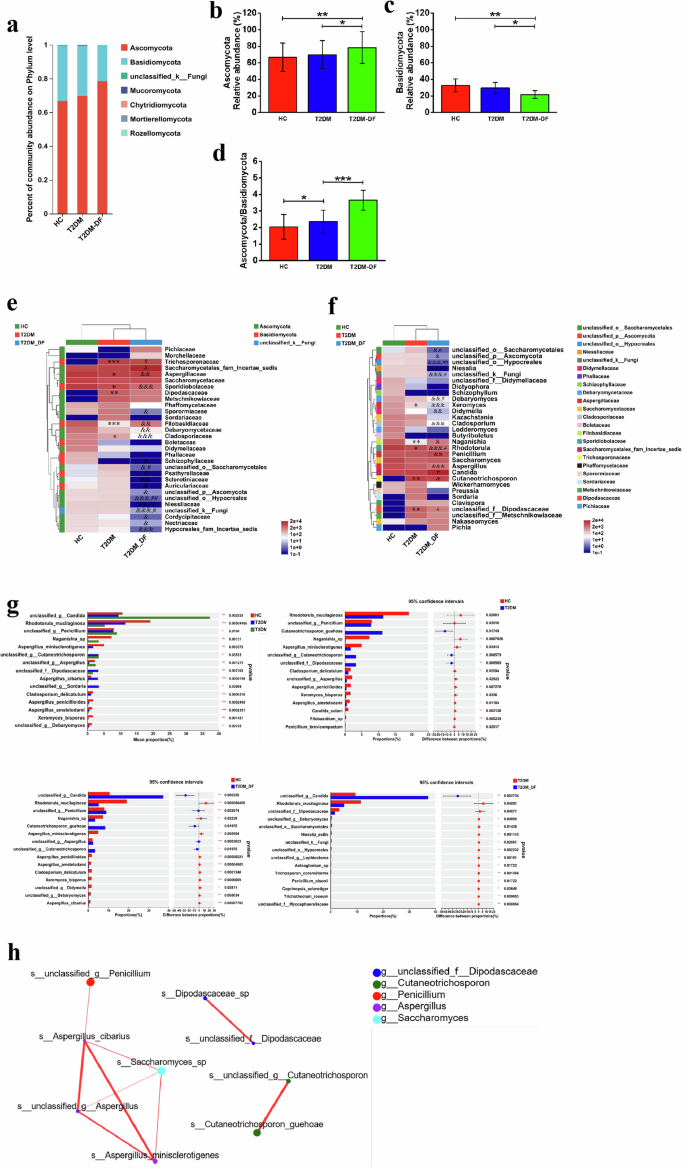
a The relative abundance of the major fungal phyla; b Relative abundance of Ascomycota; c Relative abundance of Basidiomycota; d The Ascomycota to Basidiomycota ratio; e Relative abundance of the top 30 different families; f Relative abundance of the top 30 different genera; g Relative abundance of the top 15 different species. h Network of fungi. Analyzed by Spearman rank correlation analysis. The size of the circle represents the average abundance of the species; the line represents the correlation between the two species; the thickness of the line represents the strength of the correlation; and regarding the color of the line, red represents a positive correlation. Data are expressed as means ± SEM. *p < 0.05; **p < 0.01 and ***p < 0.001 means HC versus T2DM; &p < 0.05, &&p < 0.01 and &&&p < 0.001 means HC versus T2DM_DF; #p < 0.05; ##p < 0.01 and ###p < 0.001 means T2DM versus T2DM_DF; or *p < 0.05; **p < 0.01 and ***p < 0.001.
Among the top 30 families, compared with HC, 6 families were significantly changed in T2DM, and 17 families in T2DM-DF (Fig. 2e). Among them, it was observed that Aspergillaceae, Sporidiobolaceae, Filobasidiaceae and Cladosporiaceae were significantly decreased in both T2DM and T2DM-DF, whereas Trichosporonaceae and Dipodascaceae were only drastically increased in T2DM. Sporormiaceae, Debaryomycetaceae, unclassified_o__Saccharomycetales, Sclerotiniaceae, Auriculariaceae, unclassified_p__Ascomycota, unclassified_o__Hypocreales, Niessliaceae, unclassified_k__Fungi, Cordycipitaceae, Nectriaceae and Hypocreales_fam_Incertae_sedis were only notable reduced in T2DM-DF. Remarkably, Schizophyllaceae completely disappeared in the gut of T2DM-DF (Fig. 2e). It can be seen that gut fungi of T2DM-DF were more altered at the family level. To further determine the changes, we compared gut fungi of T2DM and T2DM-DF at the family level and found that 7 families had significant changes in T2DM-DF compared with T2DM. More interesting is that Saccharomycetales_fam_Incertae_sedis alone significantly increased, while Psathyrellaceae alone significantly decreased (Fig. 2e). Therefore, these two fungi are expected to be biomarkers for the diagnosis of T2DM-DF and also provide the possibility of targeting gut fungi for the treatment of T2DM-DF.
In the top 30 genera, compared with HC, a total of 5 genera were significantly changed in the T2DM, and 15 genera in T2DM-DF (Fig. 2f). Among them, it was observed that Xeromyces, Naganishia, and Rhodotorula were significantly decreased in both T2DM and T2DM-DF, whereas unclassified_f__Dipodascaceae only drastically increased in T2DM. It’s worth noting that Cutaneotrichosporon is absent in the gut of HC, but it is significantly increased in T2DM and T2DM-DF (Fig. 2f). It reminds us that this fungus is expected to be a biomarker for diagnosing T2DM and T2DM-DF. Unclassified_o__Saccharomycetales, unclassified_p__Ascomycota, unclassified_o__Hypocreales, Niesslia, unclassified_k__Fungi, Debaryomyces, Didymella, Cladosporium, Aspergillus were only notable reduced in T2DM-DF; and Penicillium was only notable increased in T2DM-DF. Remarkably, Schizophyllum completely disappeared in the gut of T2DM-DF (Fig. 2f). It can be seen that gut fungi of T2DM-DF were more altered at the genera level. To further determine the changes, we compared gut fungi of T2DM and T2DM-DF at the genera level and found that 8 genera had significant changes in T2DM-DF compared with T2DM. More interesting is that Candida significantly increased alone, while unclassified_f__Dipodascaceae significantly decreased alone (Fig. 2f). Therefore, these two fungi are expected to be biomarkers for diagnosing T2DM-DF and also provide the possibility of targeting gut fungi for treating T2DM-DF.
Following this, an examination was conducted on the variations in the gut fungal makeup of the highest-ranking 15 species. Significant differences in the relative abundance of all 15 species were discovered among HC, T2DM, and T2DM-DF. Compared with HC, T2DM had 3 species increased, which were Cutaneotrichosporon_guehoae, unclassified_g_Cutaneotrichosporon, unclassified_f_Dipodascaceae, and 12 species decreased, which are Rhodotorula_mucilaginosa, unclassified_g_ Penicillium, Naganishia_sp, Aspergillus_minisclerotigenes, Cladosporium_delicatulum, unclassified_g_Aspergillus, Aspergillus_penicillioides, Xeromyces_bisporus, Aspergillus_amstelodami, Candida_solani, Filobasidium_sp, Penicillium_brevicompactum (Fig. 2g). Whereas in T2DM-DF, there had 5 species increased, which were unclassified_g_Candida, unclassified_g_ Penicillium, Cutaneotrichosporon_guehoae, unclassified_g_Aspergillus, unclassified_g_Cutaneotrichosporon, and 10 species decreased, which were Rhodotorula_mucilaginosa, Naganishia_sp, Aspergillus_minisclerotigenes, Aspergillus_penicillioides, Aspergillus_amstelodami, Cladosporium_delicatulum, Xeromyces_bisporus, unclassified_g_Didymella, unclassified_g_Debaryomyces, Aspergillus_cibarius (Fig. 2g). We found that some fungal species showed different trends of increases and decreases in T2DM and T2DM-DF compared to HC. To further determine the differences, we analyzed their gut fungal species. Compared with T2DM, only one species increased in T2DM-DF, which was unclassified_g_Candida, and the remaining 14 species were all reduced (Fig. 2g). It’s worth noting that unclassified_g_Candida showed a most prominent increase in T2DM-DF, but it was not in the top 15 species of HC and T2DM by the analysis.
A fungal network created by Spearman was utilized to examine the connections among these fungal species. Subsequent network analysis indicated that Aspergillus_cibarius, Saccharomyces, unclassified_g_Aspergillus, and Aspergillus_minisclerotigenes may potentially be critical players in the interactions (Fig. 2h).
Furthermore, the composition of the fungal community, which displayed significant variations between the HC, T2DM, and T2DM-DF groups, was examined using the linear discriminant analysis (LDA) effect-size method (LEfSe) (Fig. 3a, b). Then, it was figured out that taxa in different levels had differential abundance in three groups. We observed that Sporidiobolales, Microbotryomycetes, Sporidiobolaceae, Rhodotorula_mucilaginosa, Rhodotorula, Eurotiomycetes, Eurotiales, Aspergillaceae, Filobasidiaceae, Filobasidiales, Aspergillus, Naganishia_sp, Naganishia, Dothideomycetes, Aspergillus_minisclerotigenes, Agaricomycetes, Debaryomycetaceae, Capnodiales, Cladosporiaceae, Cladosporium were identified as significant biomarkers based on our observations in HC group. However, Trichosporonales, Trichosporonaceae, Cutaneotrichosporon, Sordariomycetes, Dipodascaceae, unclassified_f_Dipodascaceae, Sordaria, Sordariaceae, unclassified_g_Sordaria, Pleosporales, unclassified_f_Dipodascaceae, Aspergillus_cibarius played a significant role and could sever as biomarkers in T2DM group. Furthermore, unclassified_g_Candida, Saccharomycetales, Saccharomycetes, Ascomycota, Penicillium, unclassified_g_Penicillium, unclassified_g_Cutaneotrichosporon were essential and could serve as indicators in T2DM-DF group.
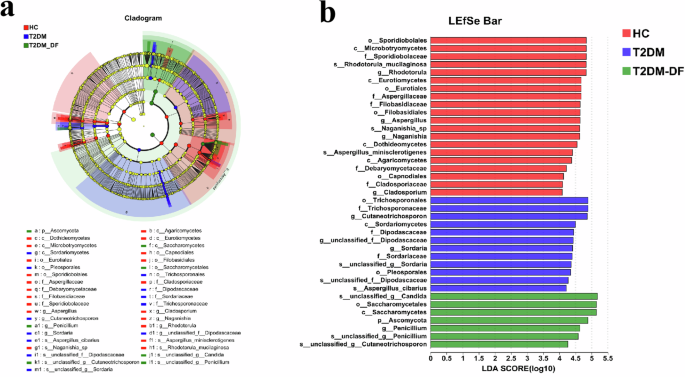
LDA effect size (LeFSe) analysis identifies discriminant taxa among the four groups. a Cladogram of the fungal community. Significant discriminant taxon nodes of the HC, T2DM, and T2DM_DF are represented by red, blue, and green, respectively. At the same time, nondiscriminant taxon nodes are represented by yellow. Branch areas are shaded according to the highest-ranked variety for that taxon. b The LDA score indicates the level of differentiation among the three groups. A threshold value of 4.0 was used as the cutoff level. Horizontal bar chart showing discriminant taxa. Significant discriminant taxa of the HC, T2DM, and T2DM_DF are represented by red, blue, and green, respectively. p < 0.05.
The gut fungal function of T2DM and T2DM-DF
Both the gut microbiota and gut fungi play crucial roles in the host’s physiological functions, as is widely recognized. Furthermore, this significant capacity impacts overall body metabolism and plays a vital role in developing endocrine disorders. Thus, PICRUSt was utilized to forecast the functional capabilities of fungi in individuals with T2DM and T2DM-DF, followed by additional examination within the Kyoto Encyclopedia of Genes and Genomes (KEGG) database framework. Subsequently, based on the results, the focus was on identifying the enzyme associated with KEGG functional categories. Compared with HC, only one enzyme had significant changes in T2DM, which was unspecific monooxygenase. This enzyme also decreased dramatically in T2DM-DF compared to HC and T2DM. Moreover, three enzymes were significantly altered in T2DM-DF alone: DNA-directed RNA polymerase and glucan 1, 4-alpha-glucosidase was notably decreased compared to HC, and histone acetyltransferase was remarkably reduced compared to HC or T2DM (Fig. 4). To sum up, the gut fungal functions had more changes in T2DM-DF than T2DM.
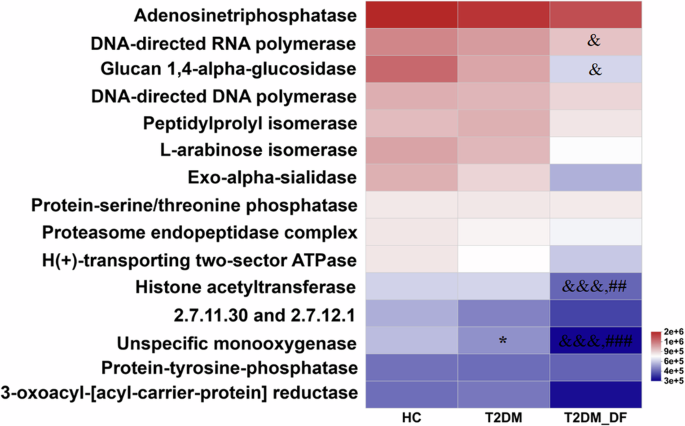
*p < 0.05; **p < 0.01 and ***p < 0.001 means HC versus T2DM; &p < 0.05, &&p < 0.01 and &&&p < 0.001 means HC versus T2DM_DF; #p < 0.05; ##p < 0.01 and ###p < 0.001 means T2DM versus T2DM_DF.
Signature fungal species in HC, T2DM, T2DM-DF biomarkers in three groups
Based on the compositional differences and functional analyses of gut fungal species in each group, and to more accurately identify the signature fungi, we used random forest to rank the top 15 importance of all fungi in each group. Results show that compared with HC, four of the top 15 most important fungal species in T2DM and T2DM-DF were the same; 11 fungal species could be used as biomarkers to distinguish T2DM or T2DM-DF from healthy individuals. In T2DM, that is Naganishia_sp, Candida_solani, Filobasidium_sp, unclassified_f_Dipodascaceae, unclassified_g_Cutaneotrichosporon, Wallemia_ichthyophaga, unclassified_g_Erythrobasidium, Trichosporon_coremiiforme, unclassified_g_Penicillium, Cystobasidium_laryngis, Mucor_racemosus (Fig. 5b). And in T2DM-DF, that is Candida_solani, unclassified_k_Fungi, Cladosporium_delicatulum,
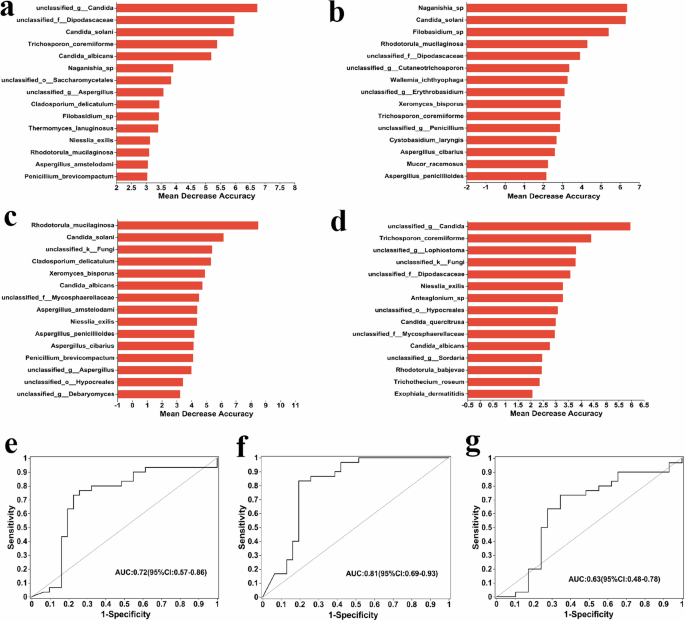
It is based on a random forest and reflects different species’ ability to affect a classification model’s accuracy. The different fungi between (a) HC versus T2DM versus T2DM_DF, (b) HC versus T2DM, (c) HC versus T2DM_DF, and (d) T2DM versus T2DM_DF. The y-axis is the importance of species, the X-axis is equal to the importance measurement value of species/standard deviation value, and The Y-axis corresponds to species names in order of importance. e–g ROC analysis of three groups: HC versus T2DM, HC versus T2DM_DF, and T2DM versus T2DM_DF. ROC analysis is often used to evaluate the performance of classification models in microbiome data, for example, to distinguish the state of samples before and after processing, determine differences between different biological samples, etc. The X-axis of Specificity is 1–0, The Y-axis is Sensitivity, and the coordinate axis is 0–1. The point marked on the curve is the optimal critical value (the values in parentheses are the corresponding Specificity and Sensitivity of the point). “bar” is the confidence interval of Specificity and Sensitivity corresponding to the point. The AUC marked in the figure is the area under the corresponding curve; The area value under the ROC curve is usually between 1.0 and 0.5. The closer the AUC is to 1, the better the diagnostic effect is. AUC has low accuracy at 0.5–0.7, sure accuracy at 0.7–0.9, and high accuracy above 0.9.
Candida_albicans, unclassified_f_Mycosphaerellaceae, Aspergillus_amstelodami, Niesslia_exilis, Penicillium_brevicompactum, unclassified_g_Aspergillus, unclassified_o_Hypocreales, unclassified_g_Debaryomyces (Fig. 5c). This result also corroborates part of the LEfSe analyses. Next, to better distinguish the differential fungal species between T2DM and T2DM-DF, we used the random forest to rank the signature distinct species between the two groups. Among the top 15 fungi, 2 were identical to T2DM, and 5 were identical to T2DM-DF when compared to HC. Hence, 8 fungi species could be used as biomarkers to distinguish DF in T2DM, they were unclassified_g_Candida, unclassified_g_Lophiostoma, Anteaglonium_sp, Candida_quercitrusa, unclassified_g_Sordaria, Rhodotorula_babjevae, Trichothecium_roseum, Exophiala_dermatitidis (Fig. 5d). Identifying these fungal species offers a foundation for forecasting the development of DF in individuals with T2DM through fungal detection techniques, potentially enabling earlier DF treatment. Finally, to further identify the signature important fungal species in the three groups, we conducted a combined analysis of three groups, which showed that 2 were not seen in the previous two-by-two comparison; there were unclassified_o_Saccharomycetales and Thermomyces_lanuginosus (Fig. 5a). The receiver operating characteristic curve (ROC) also showed a better accuracy of the predicted results (Fig. 5e–g).
Gut fungi-associated obesity index, liver function and blood parameters
The heat map correlation analysis revealed that 8 of the top 15 fungal species showed significant correlations with obesity index, liver function, and blood parameters (Fig. 6). There, 5 fungal species were significantly associated with one factor only. Aspergillus_cibarius was obviously and positively correlated with hemoglobin (Hb); Aspergillus_minisclerotigenes was obviously and positively correlated with lymphocyte (LYM); unclassified_g_Penicillium was obviously and positively correlated with blood urea nitrogen (BUN), whereas Dipodascaceae_sp was noticeably and negatively correlated with body mass index (BMI); unclassified_g_Cutaneotrichosporon was noticeably and negatively correlated with total bilirubin (TBIL) (Fig. 6). In addition, 3 fungal species were significantly associated with various factors that could potentially contribute to the onset of T2DM and T2DM-DF. Rhodotorula_mucilaginosa was dramatically and positively correlated with lymphocyte percentage (L%) but also obviously and negatively correlated with FBG and HbA1c. Naganishia_sp showed a clear and robust positive relationship with high-density lipoprotein cholesterol (HDL-C) but dramatically and negatively correlated with fasting blood glucose (FBG), glycated hemoglobin (HbA1c), and absolute neutrophil count (ANC) (Fig. 6). It means that Naganishia_sp may alleviate T2DM and T2DM-DF. unclassified_g_Candida was dramatically and positively correlated with direct bilirubin (DBIL), FBG, HbA1c, white blood cell (WBC), neutrophil granulocyte percentage (N%), and ANC, but dramatically and negatively correlated with HDL-C, L%, red blood cell (RBC) and Hb (Fig. 6). It means that unclassified_g_Candida may induce T2DM and T2DM-DF development. Overall, the results show that these 8 species of fungi are crucial in developing T2DM and T2DM-DF.
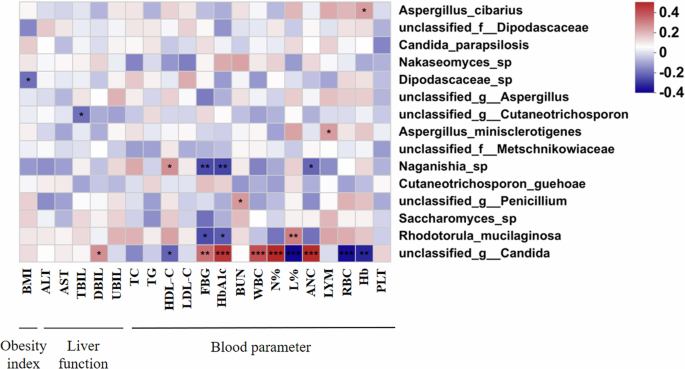
*p < 0.05; **p < 0.01; ***p < 0.001. BMI body mass index, ALT alanine aminotransferase, AST aspartate aminotransferase, TBIL total bilirubin, DBIL direct bilirubin, UBIL unconjugated bilirubin, TC total cholesterol, TG Triglyceride, HDL-C high-density lipoprotein cholesterol, LDL-C low-density lipoprotein cholesterol, FBG fasting blood glucose, HbA1c glycated hemoglobin, BUN blood urea nitrogen, WBC white blood cell, N% neutrophil granulocyte percentage, L% lymphocyte percentage, ANC absolute neutrophil count, LYM lymphocyte, RBC red blood cell, Hb hemoglobin, PLT blood platelet.
Discussion
In this study, we analyzed the fungal microbiome profiles of individuals with T2DM, T2DM-DF, and HC, with the study population not matched for gender, age, BMI, or diet. An animal study demonstrated that evolutionary forces control the emergence of mutualism between fungi and mammals [21]. The variation in fungal community composition among the three groups was evident in the richness and diversity we observed. A notable rise was noted in the abundance of Ascomycota at the phylum level in T2DM-DF compared to HC and T2DM. At the same time, a marked decline was observed in the abundance of Basidiomycota at the phylum level in T2DM-DF when compared to HC and T2DM. This means that the gut fungi in T2DM-DF were altered more drastically at the phylum level.
Ascomycota is a phylum of the kingdom Fungi. Ascomycota forms the subkingdom Dikarya with the Basidiomycota [22]. Members of the Ascomycota are commonly known as the sac fungi or Ascomycete. The members of Basidiomycota make up approximately 37% of all described species of fungi (30,000 species) and are noted for the production of large fruitbodies such as mushrooms, puffballs, brackets, etc. [23]. In a prior investigation, variations in fungal composition were observed between individuals with and without T2DM in the Emirati population. Those with T2DM showed a notable decrease in Bacteroides 2 enterotype and an increase in amino acid degradation and LPS-related modules, while non-T2DM controls had higher levels of carbohydrate degradation modules, consistent with their enterotype composition [24]. However, no study has demonstrated the gut fungal profile in patients with T2DM and T2DM-DF. The human body has an intricate and varied community of microbes, so an imbalance in the fungal mycobiome can impact the host’s health [25]. Although research into fungi is still in the beginning stages, the potential influence of fungi on endocrine disease is increasingly acknowledged.
We also found the top 15 differences in gut fungal composition at the species level between T2DM and T2DM-DF compared to HC. Both had some fungi with the same trend, while were also uniquely increased or decreased fungi, such as unclassified_g_Candida, which increased the most in T2DM-DF, and it was the only increased species compared to T2DM. Simultaneously, it was not in the top 15 species in HC and T2DM by the analysis. Therefore, unclassified_g_Candida can be used as a biomarker to predict or diagnose T2DM-DF.
Unclassified_g_Candida belonged to Candida, which means that the change in abundance of Candida may highlight the importance of gut health in susceptibility to T2DM-DF. A patient experienced worsening attention-deficit hyperactivity disorder (ADHD) and mood symptoms when infected with Candida, as reported in a case study. However, these symptoms improved after receiving effective micronutrient treatment [26]. Another study found that the abundance of Candida was higher in males with ADHD than in healthy male controls, but such a difference was not observed between females with ADHD and healthy females [27]. They also found that an in vitro permeability assay demonstrated that Candida secretion might enhance gut leakage [27]. Altered intestinal permeability indicates perturbed barrier function and has been related to the pathophysiology of T2DM and its complications. A study showed that increased zonulin levels could be a critical factor in the progression of diabetic retinopathy in individuals with type 2 diabetes [28]. The microbiome infiltrates the intestinal epithelial cells and alters the behavior of structural proteins within the cell through cytotoxins or enterotoxins [29]. Candida’s colonization of the gastrointestinal tract has been demonstrated to enhance the development of sensitivity to food allergens, partly linked to increased permeability in the gut mucosa mediated by mast cells [30]. Studies have demonstrated a correlation between high carbohydrate diets and the presence of Candida in humans. In contrast, no such correlation has been found with diets rich in amino acids, proteins, and fatty acids [17]. In a controlled feeding experiment in humans, this phenomenon was also observed in the fecal mycobiota of participants. There was an increase in Candida in subjects eating a plant-based diet while a decrease in Candida in subjects eating an animal-based diet [31]. Further investigation is required to understand whether an imbalance in gut-inhabiting yeast, such as an increase in the abundance of Candida [26], may influence optimal response and absorption of nutrients, further contributing to T2DM-DF.
The following points limit our study. Firstly, a cross-sectional design was used in this study to determine the differences in gut fungal profiles between T2DM, T2DM-DF, and HC. However, early life events and dietary patterns can influence fungal colonization and development and affect host health in later life [32, 33]. Intestinal fungal communities undergo initial colonization at birth, depending on the mode of delivery, and continue to develop during infancy, childhood, and adolescence [34]. Thus, this study cannot account for longitudinal changes and related factors associated with dysbiosis in gut fungi. Secondly, the sample sizes were smaller and had some demographic characteristics (such as age, gender, and BMI), which led to not exactly matching between samples. Although we have tried to avoid differences in disease between samples due to gender, age, and weight, our findings need to be further validated in future studies with larger sample sizes. Thirdly, although we identified intestinal fungal markers for T2DM and T2DM-DF, the molecular mechanisms underlying their interconnections with fungi, such as leaky gut phenomena or immune responses, remain unknown. Finally, using Illumina 18s rRNA sequencing technology, the identification efficiency of fungal species can reach more than 70%. However, intragenomic heterogeneity might be expected [35], so species identification of fungal communities might be incomplete or inaccurate.
Conclusion
This study explores the differential gut mycobiome profiles using Illumina 18s rRNA between patients with T2DM, T2DM-DF, and healthy control subjects. We found that the abundance of Ascomycota and Basidiomycota at the phylum level were significantly changed in T2DM and T2DM-DF, and the abundance of Candida displayed remarkable changes in T2DM-DF; they may both be identified as gut fungal markers. Simultaneously, the ROC also showed better accuracy in predicting these results.


Responses