Exploring metabolic reprogramming in esophageal cancer: the role of key enzymes in glucose, amino acid, and nucleotide pathways and targeted therapies
Introduction
Esophageal cancer (EC) is a prevalent malignancy in the digestive system. According to recent statistics, around 604,100 new cases and 544,100 deaths from EC were recorded globally in 2020 [1]. Based on the histopathological characteristics, EC is typically classified into esophageal squamous cell carcinoma (ESCC) and esophageal adenocarcinoma (EAC). Among these, ESCC is the most common, accounting for 85.79%, while EAC and other pathological types account for 11.00% and 3.21%, respectively [2]. The risk factors for EC include smoking, excessive alcohol consumption, unhealthy dietary habits (such as high salt intake and consuming very hot foods), gastroesophageal reflux disease (GERD), obesity, and exposure to radiation. Each of these factors can independently or synergistically increase the risk of EC (Fig. 1) [3, 4]. In addition to external environmental selection pressures, alterations in key genes and critical metabolic enzymes in EC cells, such as mutations or abnormal expressions in TP53 [5], PIK3CA [6], and lactate dehydrogenase (LDH) [7], can also drive the progression of EC by promoting metabolic reprogramming in cancer cells. This metabolic reprogramming allows tumor cells to adapt to harsh microenvironments, while simultaneously promoting uncontrolled proliferation, survival, and invasion, which enhances the resistance of EC to anticancer therapies.
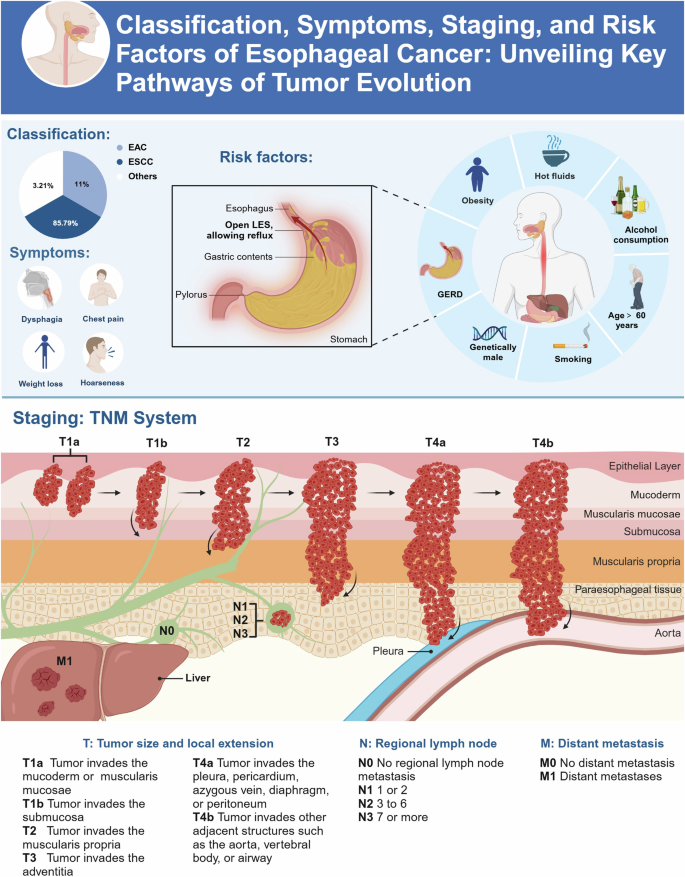
The upper section of the chart categorizes EC into ESCC, EAC, and other types, and lists typical symptoms including dysphagia, chest pain, weight loss, and hoarseness. The section on risk factors details multiple factors including GERD, smoking, alcohol consumption, intake of hot fluids, obesity, age. The lower part of the chart demonstrates the staging of EC through the TNM system, ranging from Tis (high-grade dysplasia) to T4b (invasion into structures that are unresectable), and provides detailed information on regional lymph nodes (N0 to N3) and distant metastasis (M0 and M1). Figure created with BioRender.com.
The tumor microenvironment exhibits significant differences compared to that of normal cells. Consequently, tumor cells must rapidly adapt to adverse conditions, such as nutrient deprivation and hypoxia. Metabolic reprogramming serves as a crucial energy-regulatory mechanism that enables tumor cells to survive and thrive under these hostile environmental stresses. For instance, tumor cells typically upregulate glycolysis to meet their high energy demands, a process known as the “Warburg effect”, whereas normal cells predominantly rely on oxidative phosphorylation for energy production [8]. This metabolic shift towards glycolysis not only results in the accumulation of glycolytic intermediates and lactate but also reshapes other key metabolic processes within tumor cells, such as nucleotide, amino acid, and lipid metabolism. Additionally, this process promotes the acidification of the tumor microenvironment, thereby driving tumor cell invasion and migration [9]. These metabolic alterations are driven by several key enzymes that regulate metabolism. In EC, the upregulation of specific metabolic enzymes significantly impacts tumor cell metabolic reprogramming. For example, the upregulation of hexokinase (HK) and LDH significantly activates the glycolytic process in EC cells [10, 11]. Enzymes involved in amino acid metabolism, such as glutaminase (GLS) and arginase, support the rapid proliferation of tumor cells by regulating growth factors and cell division [12, 13]. In nucleotide metabolism, the dysregulation of nucleotide synthesis enzymes, such as dihydroorotate dehydrogenase (DHODH) and ribonucleotide reductase (RNR), directly affects signaling transduction, DNA replication and repair processes that are essential for tumor growth in EC cells [14, 15]. Existing literature has reported the role of lipid metabolic reprogramming in EC [16, 17]. However, the specific mechanisms underlying the roles of glucose, amino acid, and nucleotide metabolism in the metabolic reprogramming of EC remain to be elucidated further. Comprehensive studies of these metabolic pathways can not only unveil new insights into tumor biology but also offer clues for developing therapeutic strategies that target key metabolic enzymes, paving the way for innovative cancer treatments.
In this review, we delve into the role of metabolic reprogramming in the progression of EC, with a particular emphasis on alterations in glucose, amino acid, and nucleotide metabolism pathways. We specifically focus on the aberrant changes in key metabolic enzymes and regulatory mechanisms. Additionally, we provide a comprehensive overview of current drugs targeting these enzymes, including a range of inhibitors that are currently available in clinical practice or in the developmental pipeline. Our objective is to thoroughly understand the metabolic reprogramming in EC and its potential applications in future cancer treatment strategies. Additionally, we explore possible future research directions to provide new insights and approaches for the treatment and management of EC.
EC and glucose metabolism
Compared to normal cells, glucose metabolism in cancer cells typically exhibits abnormalities. This is reflected not only in the significantly enhanced glycolytic metabolism, known as the “Warburg effect”, but also in the “reverse Warburg effect” resulting from metabolic coupling between tumor cells and cancer-associated fibroblasts in the tumor microenvironment. Cancer cells exploit multiple mechanisms to enhance glucose metabolism, suggesting that metabolic abnormalities can arise throughout various processes, including glycolysis, the TCA cycle, and the pentose phosphate pathway (PPP) (Fig. 2). In this section, we analyze the expression and activity changes of key metabolic enzymes within these three metabolic pathways, revealing the complex mechanisms by which EC cells adjust their metabolism to adapt for survival and growth.

The primary pathways of glucose metabolism include glycolysis, the TCA cycle, and the PPP. Enzymes are highlighted in red, while regulators are depicted in green. Figure created with BioRender.com.
Glycolysis
Aberrant activation of glycolysis is one of the hallmarks of EC development and progression. Unlike normal esophageal cells, transformed EC cells tend to obtain energy through the aerobic glycolysis. Several studies have shown that a variety of enzymes involve in glycolysis are significantly altered in EC (Fig. 2). Glucose transporters (GLUTs) are a family of facilitative diffusion proteins located on the cell membrane, responsible for passively transporting glucose from the extracellular environment into cells to meet their energy demands. In EC, the expression of multiple GLUTs, such as GLUT1 and GLUT3, are upregulated. Research findings reveal that the overexpression of GLUTs in ESCC may enhance tumor cell adaptability to the microenvironment by increasing glucose uptake, although the specific benefits of enhanced adaptability in EC still require further investigation [18]. Moreover, the high expression of GLUT1 is closely associated with the invasiveness and poor prognosis of ESCC, making it a potential therapeutic target [19]. EC cells enhance GLUTs expression through various mechanisms. For instance, recent studies indicate that centromere protein N (CENPN) promotes GLUT1 expression by activating the PI3K/AKT pathway, and knockdown of CENPN significantly inhibits glycolysis levels and growth in EC cells [20]. Additionally, under hypoxic conditions, lncRNA G077640 prevents the degradation of hypoxia-inducible factor 1α (HIF1α) by directly interacting with histone H2AX, thereby upregulating GLUT4 expression and further promoting the proliferation and migration of ESCC [21]. Although the relationship between GLUTs and EC has been well elucidated, clinical translation remains challenging. Excessive inhibition of GLUTs may negatively impact the energy metabolism of normal cells, making it essential to carefully balance this consideration in therapeutic design.
HK is a key enzyme in the glycolytic pathway, catalyzing the conversion of glucose to glucose-6-phosphate. There are four identified isoforms of HKs: HK1, HK2, HK3, and HK4. Notably, HK2 is highly expressed in EC patients and is significantly associated with poor prognosis [22]. The high expression of HK2 may be attributed to its ability to bind to mitochondria, thereby providing ATP more efficiently to meet the energy demands of malignant tumors [23]. EC increases the expression of the HK2 protein via multiple mechanisms. For instance, tumor cells promote the m6A modification of HK2 mRNA to increase its stability, thereby upregulating HK2 expression [11]. Additionally, overexpression of HCP5 could promote the translation efficiency of HK2 mRNA by enhancing the binding of YTHDF1 to m6A-modified mRNA [24]. Tumor cells also downregulate sirtuin-3 (SIRT3) to reduce their reliance on oxidative phosphorylation, thus promoting the upregulation and increased activity of HK2 [25]. In EC, lncRNA CASC7 has been reported to upregulate the expression of HK2 protein by inhibiting miR-143-3p, revealing a novel regulatory pathway. However, its role under normal physiological conditions and its generalizability across different cell types require further investigation [26]. These mechanisms fully demonstrate the importance of HK2 in EC glucose metabolism reprogramming. Future research should explore the interactions and synergies among these regulatory pathways to clarify the efficacy of HK2 as a therapeutic target in EC. Moreover, as a potential target for improving EC treatment whereas HK2 complex regulatory network and potential side effects must be carefully considered in clinical applications to ensure both the effectiveness and safety of therapeutic strategies.
Phosphofructokinase (PFK) is a key rate-limiting enzyme in glycolysis, responsible for catalyzing the conversion of fructose-6-phosphate (F6P) to fructose-1,6-bisphosphate (F-1,6-BP). The activity of PFK1 is precisely regulated by various metabolites. Among these, F-2,6-BP is a most potent allosteric activator of PFK1, capable of reversing ATP-mediated inhibition, allowing glycolysis to continue even in the presence of high ATP levels. PFKFB3 is a key enzyme catalyzing the production of F-2,6-BP, has been found overexpressed in various tumors, including EC [27]. In ESCC, lncRNA AGPG was found to prevent APC/C-mediated ubiquitination and degradation by directly binding to PFKFB3, thereby promoting glycolytic activity and cellular proliferation [28]. The loss of p53 in ESCC promotes significant upregulation of AGPG expression, thereby promoting ESCC progression [28, 29]. Despite the strong link between PFKFB3 and EC, targeting PFKFB3 as a therapeutic strategy presents significant challenges. The metabolic plasticity of EC cells allows them to bypass PFKFB3 inhibition by switching to other metabolic pathways. Additionally, inhibiting PFKFB3 may adversely affect glucose metabolism in normal cells, potentially leading to toxicity. These factors complicate the development of PFKFB3-targeted therapies in the treatment of EC.
Aldolase catalyzes the cleavage of the product of PFK1, F-1,6-BP, into dihydroxyacetone phosphate (DHAP) and glyceraldehyde 3-phosphate (G3P) [30]. There are three isoforms of aldolase, with abnormal expression of aldolase A and aldolase C is closely associated with the progression of EC [31]. Aldolase A known can be anchored to actin filaments in the cytoskeleton, inhibiting its catalytic activity by restricting its mobility and substrate accessibility. EC cells express LOXL2 and its splice variant L2Δ13, promoting the dissociation of aldolase A from the actin cytoskeleton and translocation into the cytoplasm, which increases substrate accessibility for aldolase A, thereby enhancing the enzyme’s capacity for reactions. Additionally, as novel deacetylases, LOXL2 and L2Δ13 directly catalyze the deacetylation of aldolase A at the K13 site, enhancing glycolysis and accelerating EC progression [32]. MiR-378a-3p, a microRNA, has been found to directly suppress aldolase A mRNA expression, thereby reducing the energy metabolism of ESCC cells and promoting early apoptosis [33]. Thus, aldolase plays a critical role in the dysregulated glycolytic metabolism in EC, and future research should exploit its potential as a therapeutic target.
Phosphoglycerate kinase 1 (PGK1) catalyzes the ATP-generating step in glycolysis, converting 1,3-bisphosphoglycerate (1,3-BPG) to 3-phosphoglycerate (3-PG). Clinical research demonstrates that PGK1 mRNA expression is significantly elevated in EC and closely correlates with the tumor’s TNM stage [34]. NRSN2-AS1, a lncRNA upregulated in ESCC tissues, has been found to directly interact with PGK1, increasing protein levels by inhibiting ubiquitination, thereby promoting glycolysis in EC cells [35]. A comprehensive multi-omics analysis involving 786 micro-tumor samples from 154 ESCC patients at various pathological stages revealed that the phosphorylation level of at the S203 site in PGK1 increases with ESCC progression, implying that PGK1 phosphorylation may be associated with tumor development. In vitro experiments demonstrated that phosphorylation of PGK1 at S203 significantly promotes the proliferation of EC tumor cells, reduces OCR and ATP production meanwhile increases ECAR, indicating an activation of glycolysis and inhibition of the citric acid cycle. This study reveals that hyperphosphorylation of PGK1 may serve as a potential drug target in the progression of ESCC [36]. In summary, PGK1 plays a crucial role in the metabolic reprogramming of glycolysis in EC and represents a potential target for EC therapeutic development.
Pyruvate kinase M2 (PKM2) is essential for regulating glycolysis in EC. Elevated levels of PKM2 have been detected in tumor tissues and plasma-derived exosomes of ESCC patients, and the expression of PKM2 is associated with increased proliferation and migration of ESCC cells in vitro, as well as poor prognosis in clinical patients [37, 38]. Findings confirm that CD276 can activate STAT3 signaling pathway, thus promoting PKM2 phosphorylation and ESCC cell proliferation. Moreover, the excessive activation of PKM2 leads to lactate accumulation, which inhibited the function of CD8+ T cells, thereby promoting the immune evasion of ESCC tumor cells [39]. Although the study highlights the role of PKM2 in immunotherapy resistance in ESCC, the limited sample size may affect the generalizability of the finding. ATP6V0C is a key regulator of intracellular and extracellular pH balance and essential for maintaining organelle acidification. Research has shown that in EC, ATP6V0C gene mutations or amplifications are common. These alterations can enhance the phosphorylation of PKM2, thereby activating its catalytic function [40]. The mechanism offers new insights into the regulation of PKM2. However, due to the broad role of ATP6V0C in cellular acidification, its dysregulation could lead to more complex systemic effects. This adds an additional layer of complexity to PKM2-targeted therapies. Estrogen-related receptors are a family of orphan nuclear receptors, including three subtypes are ESRRA (NR3B1), ESRRB (NR3B2), and ESRRG (NR3B3). Studies have shown that ESRRG expression is significantly reduced in ESCC tissues compared to adjacent non-cancerous tissues and is associated with poor prognosis in ESCC patients. Mechanistic research reveals that ESRRG binds to the PKM promoter and inhibits PKM2 protein expression. Reduced ESRRG levels significantly increase PKM2 expression and enhance ESCC cell activity. The ESRRG-specific agonist DY131 significantly inhibits glycolysis in EC cells and further validates the mechanism [41]. Nuclear factor erythroid 2-related factor 2 (Nrf2) is a key transcription factor involved in cellular responses to oxidative stress. In EC, Nrf2 promotes tumor progression by enhancing glutathione (GSH) metabolism and reactive oxygen species (ROS) detoxification, leading to adverse clinical outcomes for patients [42]. Additionally, Nrf2 can directly regulate the expression and catalytic activity of PKM2. Research has shown that activating Nrf2 can enhance the post-translational modification and tetramer formation of PKM2, which significantly promotes ESCC cell proliferation [43]. Another study showed that the expression of PKM2 is associated with chemotherapy resistance in ESCC patients. Inhibiting PKM2 can restore ESCC patient sensitivity to cisplatin, thereby enhancing therapeutic efficacy. This study suggests a potential strategy for targeting PKM2 to overcome chemotherapy resistance in ESCC [44]. Additionally, research reports indicate that environmental cadmium may induce EC by upregulating Cyclin-dependent kinase 6 (CDK6) expression, which subsequently activates excessive phosphorylation of PKM2 and inhibits its pyruvate kinase activity. This inhibition redirects glucose-derived carbon into the PPP, promoting the production of NADPH and GSH to counteract ROS, thereby suppressing EC cells apoptosis [45]. Although PKM2 holds significant potential for anti-EC therapy, its diverse biological functions within cells suggest that broad inhibition may trigger a range of unintended physiological responses. These effects could lead to unpredictable therapeutic outcomes and might even promote tumor resistance or recurrence. Therefore, the use of PKM2 inhibitors in EC treatment should be caution.
LDHA is a key enzyme in cellular energy metabolism, particularly under hypoxic conditions in normal cells and in certain cancers that heavily rely on glycolysis for energy production, such as breast cancer, colorectal cancer and EC [46]. In these tumors, the upregulated activity of LDHA not only supports the high metabolic demands of cancer cells by converting large amounts of pyruvate into lactate but also suppresses immune cell activity and increases the invasiveness of cancer cells by altering the tumor microenvironment through enhanced lactate accumulation. In clinical samples of ESCC, LDHA expression was found to be significantly upregulated, leading to the activation of the AKT signaling pathway, increased cyclin D1 levels, and the promotion of ESCC progression [47]. Additionally, TNF-α can induce the gene expression of LDHA and LDHB, thereby increasing lactate secretion by EC cells. Inhibition of LDHA activity can reduce TNF-α induced migration and the expression of MMP9 in EC cells, thus modulating their survival and invasive capacity [48]. Lactate is not only an important metabolic byproduct but also promotes the further progression of cancer by acidifying the tumor microenvironment. Recent studies have revealed a novel regulatory mechanism that circLPAR3 regulates LDHA by sequestering miR-873-5p, thereby enhancing tumor growth, metastasis, and glycolytic activity in ESCC [49]. Although LDHA activity is closely linked to the tumor microenvironment and immune evasion, most existing studies are based on in vitro experiments, lacking sufficient clinical evidence to support its role across different EC subtypes. Moreover, given the broad involvement in various physiological and pathological processes of LDHA, its application in EC treatment requires further targeted investigation.
Pyruvate Dehydrogenase Kinase (PDK) is an enzyme that regulates the activity of the Pyruvate Dehydrogenase Complex (PDC). In tumor cells, PDK promotes the phosphorylation of Pyruvate Dehydrogenase (PDH), a key enzyme of the PDC, reducing the conversion of pyruvate to Acetyl-CoA and promoting energy acquisition via glycolysis instead of oxidative phosphorylation [50]. In EC, the upregulation of PDK1 enhances glycolysis and promotes tumor cell growth and survival. Conversely, reducing the expression or activity of PDK1 can promote cell apoptosis. A specific miRNA that targets PDK1, miR-6516-5p, can directly suppress PDK1 expression and shows a synergistic effect with cisplatin in inhibiting ESCC cells in both in vitro and in vivo models [51]. Circ_0001944 was reported to enhance PDK1 expression by directly binding to miR-338-5p, a PDK1-specific miRNA, thereby promoting glycolysis in EC cells [52]. Another LncRNA, protein tyrosine phosphatase receptor G antisense RNA 1 (PTPRG-AS1), enhances PDK1 expression by sponging miR-599, thereby promoting proliferation, migration, glycolysis, and stem-like characteristics in ESCC cells [53]. These studies underscore the pivotal role of PDK1 in regulating metabolic reprogramming and tumor progression in EC. The non-coding RNA further demonstrates the complexity of PDK1 regulation. These findings suggest that targeting PDK1, either directly or through the modulation of its regulatory networks, could be a promising strategy for improving EC treatment outcomes. However, further research is needed to explore the full therapeutic potential of PDK1 inhibition across different EC subtypes and to assess its impact on long-term tumor suppression.
TCA cycle
The TCA cycle, located in the mitochondria, is a key pathway in cellular metabolism that drives the conversion of nutrients into ATP. Its metabolic intermediates and products, such as NADH, are involved in various cellular functions and are critical for cell survival and proliferation. TCA cycle exhibits adaptability through its numerous reversible reactions, allowing for dynamic regulation of metabolic flux to support cells in adapting to fluctuating nutrient and energy demands. Dysregulation of this cycle is commonly observed in various cancers, including EC, highlighting its critical role in cellular metabolism and disease progression. The relationship between the TCA cycle and metabolic reprogramming in EC is illustrated in Fig. 2.
Isocitrate dehydrogenase (IDH) is a key enzyme in the TCA cycle that catalyzes the oxidative decarboxylation of isocitrate to produce α-ketoglutarate (α-KG). Extensive research has shown that mutations in the IDHs gene are closely associated with various types of cancer, such as gliomas, acute myeloid leukemia (AML), and soft tissue sarcomas [54]. Point mutations in IDHs alter the substrate specificity, causing it to convert α-KG into 2-hydroxyglutarate (2-HG). Excessive 2-HG disturbs cellular metabolism and epigenetic regulation, thereby promoting tumorigenesis. However, researchers did not find any mutations in IDH1 or IDH2 when investigating the incidence of IDHs mutations in 10 ESCC cell lines and 96 ESCC tissue samples [55]. Some cases of ESCC exhibited elevated levels of 2-HG without IDH1 or IDH2 mutations, suggesting that these mutations play a limited role in the development of ESCC. The high levels of 2-HG may be independent of IDHs mutations and could be associated with ESCC progression. Although no IDHs mutations have been identified in ESCC, it is reported that the expression levels of IDH2 are upregulated in ESCC and may serve as a valuable biomarker for assessing the prognosis of ESCC patients [56]. Moreover, clinical data confirm that the expression level of IDH2 is associated with the effectiveness of radiotherapy in ESCC patients. Mechanistic research suggests that IDH2 promotes radiosensitivity by inhibiting AKT phosphorylation in a ROS-dependent manner. Additionally, in mouse xenograft tumor models, the loss of IDH2 was found to enhance radiation-induced growth inhibition and apoptosis. Therefore, targeting IDH2 may represent a promising strategy to improve the efficacy of radiotherapy in ESCC patients [57].
ATP citrate lyase (ACLY) cleaves citrate, derived from the TCA cycle, into acetyl-CoA and oxaloacetate in the cytoplasm. Acetyl-CoA is a key precursor for synthetic pathways such as fatty acid and cholesterol biosynthesis. Oxaloacetate can be further converted into pyruvate, which re-enters the TCA cycle, completing the metabolic loop. Recent studies indicate that ACLY activity is closely associated with the proliferative capacity of EAC cells [58]. Moreover, SIRT2, as a deacetylase, can enhance ACLY activity by inhibiting its acetylation, thereby promoting lipid metabolism in ESCC cells. The SIRT2 inhibitor AGK2, by increasing ACLY acetylation levels, suppresses ESCC cell proliferation and migration [59]. Furthermore, evidence suggests that the m6A reader HNRNPA2B1 promotes EC cell progression by upregulating ACLY, thereby serving as a potential oncogenic factor [60]. These findings highlight the critical regulatory role of ACLY in the biological behavior of EC cells and suggest its potential as a therapeutic target for EC treatment.
PPP
To meet the increasing growth demands of cancer cells, the PPP is typically upregulated to supply essential building blocks and redox cofactors, such as NADPH and GSH. This upregulation of the PPP in tumor involves the regulation of glucose-6-phosphate dehydrogenase (G6PD), a key enzyme that limits the rate of PPP (Fig. 2) [61]. Research indicates that the high expression of G6PD in ESCC is closely correlated with histological grading, lymph node metastasis, and poor prognosis [62]. Although elevated G6PD expression is associated with EC progression, it as a prognostic biomarker remains to be investigated, particularly given the potential differences in its expression across various EC subtypes and pathological stages. G6PD activity is regulated by glycosylation modification. Investigations have revealed that Erk-O-GlcNAc transferase (OGT) promotes G6PD activation by enhancing its O-GlcNAcylation. The expression of OGT is regulated by the DNA polymerase iota (Pol ι) activated Erk signaling pathway. This post-translational modification enables G6PD to more efficiently channel glucose into the PPP, increasing NADPH production, which boosts the metabolic activity of tumor cells and promotes ESCC proliferation [63]. In a study investigating the pathogenesis of human EAC using a rat duodenal reflux model, researchers found that esophageal cells exposed to bile acids over an extended period showed significantly elevated expression of OGT and G6PD, along with activation of the PPP, which in turn promoted the occurrence of EAC [64]. Although the reflux model revealed potential mechanisms by which bile acid reflux contributes to EAC, the heterogeneity between the model and human EAC pathology may limit the extrapolation of the finding. Polo-like kinase 1 (PLK1), a protein involved in cell cycle regulation, was reported to activate G6PD by promoting its dimerization, leading to increased levels of NADPH and GSH. This inhibits ferroptosis in ESCC cells, thereby reducing their sensitivity to radiotherapy and chemotherapy [65]. In summary, the upregulation of the PPP through G6PD activation plays a critical role in supporting the metabolic demands of EC cells, driving proliferation and resistance to therapies. The involvement of glycosylation modification and signaling pathways, further highlights the complexity of this regulation. While experimental models, like the bile acid reflux rat model providing valuable insights into the pathogenesis of EC, their limitations in fully replicating human disease underscore the need for further clinical validation.
Transketolase (TKT) is an enzyme in the non-oxidative branch of the PPP, involved in converting ribulose-5-phosphate (Ru-5-P) into F6P and glyceraldehyde-3-phosphate (G3P). This process contributes to the regulation of glucose metabolism and nucleotide synthesis in cells. In tumors, TKTL1, a subtype of TKT, can enhance glycolysis and glutamine metabolism by forming a dimer, thus sustaining the high proliferative state of tumor cells [66]. Recent studies indicate that the expression of TKTL1 is significantly elevated in ESCC tissues and cells. Inhibition of TKTL1 expression using siRNA can markedly reduce the proliferation and metastatic potential of ESCC cells [67]. Mechanism research found that knockdown of TKTL1 leads to significant cell cycle arrest in the G0/G1 phase in vitro. During this process, decreased levels of cyclin D1 and CDK4 were observed, along with a reduction in the Ki67 proliferation index in ESCC cells, confirming the importance of TKTL1 in regulating tumor cell proliferation and survival [68]. Although the impact of TKTL1 inhibition on the cell cycle and proliferation suggests the potential therapeutic value in EC, current research remains largely confined to cellular and animal models, with a lack of clinical validation. Future research should focus on its potential as biomarker for EC and further evaluate its efficacy and safety as EC target.
Metabolism of amino acids in EC
Cancer cells frequently augment the uptake of essential amino acids and enhance the synthesis of non-essential amino acids to fulfill their requirements for protein synthesis, signal transduction, antioxidation, and energy generation. They display considerable metabolic adaptability, allowing them to thrive even in environments where amino acid availability is restricted. Moreover, these cells exhibit substantial metabolic heterogeneity within their microenvironment. In EC, this reprogramming often causes fluctuations in tissue or plasma amino acid levels [69]. For instance, there are significant differences in cysteine levels between EC tissues and adjacent tissues [70]. Additionally, multiple amino acids, such as aspartate, glutamate, histidine, serine, and proline, are significantly reduced in the plasma of EC patients, while nitric oxide levels and arginase activity are increased [71, 72]. The synthesis of certain non-essential amino acids is particularly crucial in EC, for example, the pathway for methionine synthesis is frequently activated, promoting tumor cell proliferation and correlating with tumor invasiveness and drug resistance. Thus, the detailed relationship between amino acid metabolism and the progression of EC is depicted in Fig. 3.

The significantly altered pathways include glutamine metabolism, tryptophan metabolism, serine-glycine and one-carbon metabolism, as well as arginine metabolism. Red indicates key enzymes, while green indicates regulators of the key enzymes. Figure created with BioRender.com.
Glutamine metabolism
The glutamine metabolism pathway is frequently activated in EC, indicating that glutamine is not only a vital nutrient for cell survival but also a key factor in regulating cancer cell growth and proliferation. In this process, glutamine transporters, particularly solute carrier family A1 member 5 (SLC1A5), play a central role in maintaining intracellular glutamine levels and regulating cellular glutamine metabolism. Clinical evidence shows that the expression of SLC1A5 is significantly upregulated in EC. Knockdown of SLC1A5 markedly inhibits the uptake of glutamine and leucine by EC cells, suppresses the mTORC1 signaling pathway and promotes apoptosis in EC cells [73]. Additionally, some circular RNAs were found to modulate SLC1A5 expression by acting as microRNA sponges, thereby affecting glutamine metabolism. For instance, circ_0001273 can inhibit glutamine metabolism in EC cells by targeting the miR-622/SLC1A5 signaling axis, while circ-SFMBT2 binds to miR-107, enhancing SLC1A5 expression and promoting the malignant progression of EC [74, 75]. In conclusion, SLC1A5 may be a potential target for the treatment of EC, although there is currently a lack of clinical validation evidence.
GLS is a key enzyme in glutamine metabolism, catalyzing the conversion of glutamine into glutamate and ammonia. Clinical data confirm that GLS expression is significantly elevated in ESCC, and further mechanistic research reveals that its interaction with PDK1 within the mitochondria increases the phosphorylation levels of PDHA1, thereby inhibiting PDH activity, suppressing oxidative phosphorylation, and ultimately promoting glycolysis in ESCC [13]. RNA binding motif 4 (RBM4) is a multifunctional RNA-binding protein primarily involved in regulating various RNA metabolic processes, such as alternative mRNA splicing, transcription, and translation. Clinical and experimental data suggest that RBM4 is significantly expressed in ESCC, and mechanistic studies have revealed that it activates glutamine metabolism by inhibiting the LKB1-AMPK axis, thereby bypassing senescence and sustaining cell proliferation. Treatment with the GLS inhibitor CB-839 was found to significantly suppress RBM4-overexpressing ESCC tumors. This study highlights the potential of targeting GLS as a therapeutic strategy for ESCC with high RBM4 expression [76]. Clinical data confirm that dysregulation of the Fbxo4-cyclin D1 axis occurs frequently in ESCC. Further studies have revealed that Fbxo4 mutations or cyclin D1 overexpression drive Gln-addiction in ESCC cells, increasing their sensitivity to the GLS inhibitor CB-839. This research suggests that GLS plays a central role in the Fbxo4-cyclin D1 axis-driven progression of ESCC, indicating that CB-839 could be a potential therapeutic agent for treating ESCC [77]. Circ_0001093, a circular RNA, functions as a competitive endogenous RNA by sponging miR-579-3p, thereby increasing GLS expression and promoting glutamate metabolism [78]. Overall, GLS may serve as a therapeutic target for specific subtypes of EC characterized by RBM4 overexpression or Fbxo4 mutations. CB-839 has emerged as a potential treatment although further clinical data are required to substantiate its efficacy.
Glutamate dehydrogenase (GDH) converts glutamate into α-KG and ammonia, a critical step in glutaminolysis. The α-KG then enters the TCA cycle, providing energy for the cell. Research has shown that the catalytic activity of GDH is inhibited by SIRT4 through the addition of ADP-ribose groups to the GDH protein. By inhibiting GDH, SIRT4 exerts a negative feedback effect on glutamine metabolism and energy production. This regulatory mechanism ensures that cells do not overconsume glutamine when energy is abundant, and helps maintain metabolic balance when cells need to conserve resources or respond to stress [79]. Dysregulation of the SIRT4-GDH axis has been closely linked to the progression of various tumors. Currently, studies on the role of GDH in EC are limited. However, some studies have found that SIRT4 expression is downregulated in ESCC tumor tissues, and knocking down SIRT4 significantly increases GDH activity, promoting the proliferation and migration of ESCC cells [80]. Therefore, the SIRT4-GDH axis may offer new therapeutic opportunities for targeting EC, though further mechanistic studies are required to validate this potential.
Tryptophan metabolism
Tryptophan is an essential amino acid for the human body, and its metabolism primarily proceeds through two major pathways. In the first pathway, tryptophan is converted to 5-hydroxytryptophan by the action of tryptophan hydroxylase and aromatic amino acid transaminase, which is subsequently transformed into 5-hydroxytryptamine. In the second pathway, tryptophan is metabolized via the kynurenine (KYN) pathway, resulting in the production of various KYN derivatives. The KYN pathway is the predominant route of tryptophan metabolism, converting L-tryptophan to N-formyl-L-kynurenine (NFK), catalyzed by the rate-limiting enzymes indoleamine 2,3-dioxygenase (IDO) and tryptophan 2,3-dioxygenase (TDO). These metabolic products play critical roles in immune regulation [81].
Clinical investigations have shown that the expression level of IDO1 is significantly elevated in ESCC patients after chemotherapy and is associated with poor pathological response and unfavorable prognosis [82]. A study revealed that IDO1 facilitates NF-κB nuclear translocation, enhancing its binding to the CXCL10 promoter and thereby upregulating CXCL10 expression in EC cells, which promotes tumor cell metastasis and immune evasion [83]. TDO is another enzyme that catalyzes the conversion of tryptophan to KYN. Unlike IDO, which plays a key role in immune regulation, TDO primarily regulates systemic tryptophan metabolism and homeostasis. Clinical findings reveal that TDO2 is overexpressed in ESCC tissues and is significantly associated with lymph node metastasis, advanced clinical stage, and poor prognosis. Experimental data suggest that TDO2 can upregulate Interleukin-8(IL-8) expression through the AKT/GSK3β pathway, promoting M2 polarization of macrophages and suppressing the anti-tumor immune response in ESCC [84]. The enhanced expression of IDO1 and TDO2 in EC cells catalyzes the excessive production of KYN, which activates the aryl hydrocarbon receptor (AhR), promotes the generation of immunosuppressive dendritic cells and regulatory T cells, and thereby facilitates tumor immune evasion [85]. Consequently, targeting the IDO1/TDO2-KYN-AhR signaling pathway may represent a novel strategy for cancer therapy.
Serine/glycine and one-carbon metabolism
The metabolism of serine and glycine provides one-carbon units essential for the synthesis of various biomacromolecules and the maintenance of redox balance. This metabolic process is critical for sustaining essential biochemical reactions and promoting the rapid proliferation of tumor cells [86, 87]. Serine, glycine, and their related enzymes serve as one-carbon donors in the folate cycle, playing key roles in nucleotide synthesis, DNA and protein methylation, and the maintenance of redox balance, which promotes EC progression.
Serine Hydroxymethyltransferase 2 (SHMT2) is a mitochondrial enzyme that catalyzes the conversion of serine to glycine while simultaneously transferring a hydroxymethyl group to tetrahydrofolate (THF), generating 5,10-methylene-THF. This mechanism is essential for in one-carbon metabolism and is a key reaction within the cellular folate cycle. A study on the function of SHMT2 in EC revealed that its expression and lactylation modification were significantly increased under hypoxic conditions, promoting the proliferation, migration, and glycolytic activity of EC cells. Additionally, the upregulation of SHMT2 is associated with enhanced tumor stem cell properties, contributing to tumor self-renewal and pluripotency. Mechanistic research revealed that SHMT2 interacts with MTHFD1L, potentially promoting MTHFD1L expression or stability, thereby further enhancing the metabolic and survival capabilities of EC cells [88]. MTHFD1L is also a key enzyme in the mitochondrial one-carbon metabolism pathway, and the one-carbon units produced in conjunction with SHMT2 are essential precursors for GSH synthesis. This process plays a critical role in regulating ferroptosis in tumor cells. CRISPR perturbation screening revealed that the loss of SHMT2 and MTHFD1L genes significantly increases the sensitivity of EC cells to Eprenetapopt, thereby enhancing its efficacy in inducing ferroptosis in these cells [89]. In addition, the overexpression of SHMT2 in EC cells promotes the accumulation of the methyl donor SAM, thereby enhancing the m6A modification and stability of c-myc mRNA through a METTL3/FTO/ALKBH5/IGF2BP2-dependent mechanism, which promotes the malignant progression and immune evasion of EC [90]. These findings suggest that SHMT2 may be a potential target for the treatment of EC. Future studies should explore the efficacy and safety of SHMT2 inhibitors in EC.
Phosphoglycerate dehydrogenase (PHGDH) catalyzes the initial step of the de novo serine synthesis pathway, acting as a key enzyme in this metabolic pathway and serving as a bypass of glycolysis. Clinical findings reveal that its high expression in ESCC is associated with poor patient prognosis. By promoting serine synthesis and activating the Wnt/β-catenin signaling pathway, PHGDH significantly enhances the proliferation and angiogenesis of ESCC cells, underscoring its crucial role in metabolic reprogramming of ESCC and the potential as a therapeutic target [91].
Phosphoserine aminotransferase 1 (PSAT1) is considered a key transaminase linking the glycolysis pathway with serine metabolism. In EC, PSAT1 converts glutamate to 3-phosphohydroxy-pyruvate, contributing to tumor aggressiveness. It has been established that lncRNA maternally expressed gene 3 (MEG3) significantly inhibits ESCC cell proliferation, migration, invasion, and epithelial-mesenchymal transition (EMT) by suppressing the PSAT1-dependent GSK-3β/Snail signaling pathway [92]. This study suggests that PSAT1 may play a role in the EMT of ESCC, but the detailed molecular basis of this mechanism and its role in ESCC may require further comprehensive investigation. Additionally, microRNA-365 has been reported to inhibit tumor invasion and progression in ESCC by regulating PSAT1 expression [93]. Overall, the role of PSAT1 and its regulatory factors in EC warrants further investigation, with the aim of identifying novel strategies and targets for cancer treatment.
Arginine metabolism
Arginine is an essential amino acid required for the rapid growth of tumor cells. Tumor cells depend on arginine as a growth factor to promote intracellular protein synthesis, nitrogen metabolism, and cell proliferation. Some tumors, including hepatocellular carcinoma and prostate cancer, enhance arginine metabolism by upregulating arginine-related metabolic enzymes, such as arginase or nitric oxide synthase (NOS), to support rapid proliferation [94]. Data from clinical studies point to the fact that the activity of arginase and the level of nitric oxide (NO) are significantly elevated in the plasma of EC patients [95, 96]. Mechanism analyses uncovered that arginase depletes L-arginine in the tumor microenvironment by hydrolyzing L-arginine into ornithine and urea, thereby inhibiting T-cell function and promoting the growth of EC [95]. NOS is another key enzyme in the arginine metabolic pathway. Based on its expression and function, NOS is categorized into three major isoenzymes: inducible nitric oxide synthase (iNOS), endothelial nitric oxide synthase (eNOS), and neuronal nitric oxide synthase (nNOS) [97]. Data indicate that iNOS expression is significantly elevated in ESCC, leading to increased NO production and promotes cancer progression through various mechanisms [98]. For example, NO produced by iNOS enhances angiogenesis and promotes tumor cell proliferation and survival by activating cGMP pathway-related signaling. Moreover, high concentrations of NO suppress T-cell function, weakening the immune system’s ability to eliminate tumors. Additionally, NO increases the invasive potential, motility, and distant metastatic capability of tumor cells by regulating the activity of matrix metalloproteinases (MMPs) [99, 100]. Although existing studies suggest that iNOS may be an effective target for the treatment of EC, inhibiting iNOS could impair immune system function, increasing the risk of infections and other complications. Therefore, in the treatment of EC, it is essential to carefully balance the inhibition of iNOS with the maintenance of immune function.
In the urea cycle, argininosuccinate synthase 1 (ASS1) and argininosuccinate lyase (ASL) work together to convert aspartate and citrulline into arginine. This process is a crucial step in the urea cycle, as it not only maintains the continuity of the cycle by regenerating arginine but also provides substrates for various metabolic processes. Research findings reveal that in primary ESCC, the levels of ASS1 and ASL are elevated, while they are reduced in lymphatically metastatic tumor tissues. Investigations into the mechanisms revealed that Forkhead box O3a (FOXO3a) regulates the expression of ASS1 and ASL at different stages of ESCC. In primary tumors, FOXO3a is typically inactivated, promoting the transcription of ASS1 and ASL, which facilitates tumor dissemination and metastasis. Conversely, in metastatic tumors, the activation of FOXO3a suppresses the expression of ASS1 and ASL [101]. This metastatic reprogramming reflects the important roles of ASS1 and ASL in the process of ESCC metastasis. However, this study did not explain the reasons for the significant changes in the regulatory mechanisms of FOXO3a during the different stages of ESCC metastasis. Moreover, Clinical findings reveal that chemotherapy resistance is closely associated with the upregulation of ASS1 expression. Mechanistic investigations suggested that ESCC promotes the expression of ASS1 and pyrroline-5-carboxylate reductase 1 (PYCR1) through the upregulation of insulin-like growth factor-I receptor (IGF1R) expression, which facilitates the phosphorylation of c-MYC, thereby enhancing chemotherapy resistance in ESCC [102]. Therefore, future research should focus on the regulatory mechanisms of ASS1 to develop more effective therapeutic strategies to overcome resistance issues in EC.
The metabolic product of arginine, ornithine, is converted into polyamines under the catalysis of ornithine decarboxylase (ODC), a process closely related to cell proliferation that promotes tumor initiation and progression. Clinical studies indicate that ODC expression levels are significantly elevated in ESCC tissues. Inhibition of ODC activity can reduce ESCC cell proliferation, induce apoptosis, and trigger G2/M phase arrest, suggesting that ODC may be a novel therapeutic target for ESCC [103]. Additionally, ODC regulates the expression of various signaling proteins, including p38α, ERK1/2, and AKT/mTOR/p70S6K, through its metabolic product polyamines, thereby promoting the progression of precancerous lesions in ESCC [104]. Furthermore, findings confirm that the excessive production of polyamines is associated with the polarization of macrophages in the tumor microenvironment. IL-33 in ESCC cells enhances the expression of ODC and the production of polyamines, which promotes the M2 polarization of tumor-associated macrophages, reduces the tumor immune response, and facilitates the growth and metastasis of ESCC [105]. These studies suggest the important role of ODC in anti-tumor immunotherapy for EC. Future research should focus more on the role of polyamines in EC, further elucidating the specific mechanisms of ODC in the progression of the disease.
EC and nucleotide metabolism
Nucleotides, fundamental molecules of life, play essential roles in processes such as DNA and RNA synthesis, energy conversion, and signal transduction. In healthy cells, the production and utilization of nucleotides are tightly regulated to maintain cellular functions and life processes. However, this balance is disrupted in the context of cancer. Cancer cells accelerate nucleotide synthesis through endogenous mechanisms to meet their demands for rapid division and proliferation. In EC, the activities of key enzymes and metabolic pathways associated with nucleotide biosynthesis frequently undergo cancer-specific alterations, reflecting metabolic changes that drive tumor progression (Fig. 4). These changes not only lead to the excessive production of nucleotides but also disrupt the balance of cellular metabolites, further promoting cancer progression.

This figure primarily involves the key enzymes in purine and pyrimidine metabolism, as well as DNA damage repair. Red indicates key enzymes, while green denotes regulators of the key enzyme. Figure created with BioRender.com.
Thymidylate synthase (TS) is an enzyme that promotes DNA synthesis by converting deoxyuridine monophosphate into deoxythymidine monophosphate. Clinical observations support the idea that the polymorphism in the 5′-UTR of the TS gene and the expression levels of TS mRNA are significantly associated with the response to 5-fluorouracil (5-FU) treatment and prognosis in EAC patients. Specifically, EAC patients with the 3RG sequence in the 5′-UTR of the TS gene and high TS mRNA expression are less likely to benefit from 5-FU chemotherapy [106]. Moreover, mechanistic studies demonstrated that DNA binding inhibitor 1 (Id1) competitively binds to Cdc20, preventing the degradation of the E2F1 protein and enhancing its stability. The stabilized E2F1 transcription factor subsequently upregulates the expression of TS, contributing to increased resistance of EC cells to the chemotherapeutic drug 5-FU [107]. Although TS-targeted therapy for EC is relatively well-established, the issue of drug resistance remains a major clinical challenge. Elucidating the mechanisms underlying this resistance may offer new insights for improving TS-targeted therapies and enhancing treatment efficacy.
CD73 is a membrane-bound nucleotide enzyme that hydrolyzes extracellular adenosine monophosphate (AMP) into adenosine through dephosphorylation, playing a role in regulating the adenosine pathway. Adenosine is an immunosuppressive molecule that weakens the body’s anti-tumor immune response by inhibiting the activity of T cells, natural killer (NK) cells, and dendritic cells, aiding tumor cells in evading immune surveillance. Research has shown that elevated expression of CD73 is associated with decreased disease-free survival and overall survival rates in ESCC patients, significantly impacting the effectiveness of immunotherapy [108]. CD73 may be a promising immunotherapy target in EC. Developing effective CD73 inhibitors and combining them with existing immunotherapy approaches could represent a valuable treatment strategy. The high expression of the nucleotidase CD39 also affects the progression of ESCC. Evidence suggests that CD39-expressing CD8+ T cells may serve as potential molecular markers for diagnosing and predicting therapeutic outcomes in ESCC [109]. Dihydropyrimidine dehydrogenase (DPD) primarily participates in the breakdown of pyrimidine drugs and endogenous pyrimidine nucleotides. Clinical evidence shows that elevated DPD expression in locally advanced EC may serve as a potential biomarker for predicting the response to neoadjuvant therapy [110]. These findings suggest that several enzymes involved in nucleotide metabolism, including CD73, CD39, and DPD, play crucial roles in chemotherapy resistance, tumor progression, and immune evasion in EC. The expression levels and activities of these enzymes are vital for determining the prognosis of EC. Future research should focus on elucidating the mechanisms by which CD73, CD39, and DPD contribute to EC development and explore their therapeutic potential in the treatment of EC.
MutT homolog 1 (MTH1) is a phosphodiesterase that acts on oxidized nucleotides, capable of hydrolyzing 8-oxoguanosine triphosphate (8-oxo-dGTP) to prevent its incorporation into nuclear and mitochondrial DNA, reducing cytotoxicity in tumor cells [111]. Studies have demonstrated that MTH1 mRNA levels are elevated in cancerous tissues compared to normal epithelial tissues. Moreover, MTH1 expression is significantly correlated with tumor invasion depth, venous invasion, advanced cancer stages, and poorer overall and disease-specific survival rates. Consequently, MTH1 expression serves as an independent predictor of poor prognosis in EC, making MTH1 a potential therapeutic target for ESCC patients [112]. Ras guanine nucleotide-releasing protein 3 (RasGRP3) is a member of the Ras guanine nucleotide-releasing protein family, which facilitates the activation of the Ras signaling pathway by regulating the activity of small Ras family GTPases. Scientific investigations indicate that RasGRP3 is highly expressed in ESCC cells. Mechanistic investigations indicate that RasGRP3 influences the viability, migration, invasion, and apoptosis of ESCC cells by activating the PTEN/PI3K/Akt signaling pathway [113]. The activity of RasGRP3 is dependent on nucleotide metabolism, particularly the utilization of GTP, making it a potential therapeutic target for inhibiting ESCC. 8-oxoguanine DNA glycosylase (OGG1) is a widely expressed protein that initiates the base excision repair pathway to repair chemotherapy-related 8-oxoguanine damage. Overexpression of OGG1 can protect ESCC cells from cisplatin-induced apoptosis and prolong the survival of cancer cells [114]. OGG1 plays a critical role in the chemoresistance of EC, suggesting that it could serve as a target for chemosensitization. In conclusion, these findings indicate that MTH1, RasGRP3, and OGG1 are key proteins involved in the regulation of nucleotide metabolism and represent potential therapeutic targets for EC. Future studies should focus on exploring the potential application of specific inhibitors targeting these enzymes in EC treatment.
DHODH is a rate-limiting enzyme in the pyrimidine biosynthesis pathway, providing essential precursors for nucleic acid synthesis in cells. Experimental data suggest that DHODH significantly promotes the proliferation of EC cells and inhibits ferroptosis [115]. Furthermore, DHODH directly binds to the NH2 terminus of β-catenin, preventing its ubiquitin-mediated degradation by disrupting the interaction with GSK3, leading to its accumulation in the nucleus. This nuclear accumulation of β-catenin activates downstream gene expression, promoting the proliferation of ESCC cells [15]. DHODH is a well-established therapeutic target with clinically developed drugs. Future research should focus on evaluating the safety and efficacy of these drugs in the treatment of EC. RNR plays a critical regulatory role in DNA synthesis. Ribonucleotide reductase subunit M1 (RRM1) is a component of RNR, and evidence suggests that in ESCC RRM1 can inhibit glucose degradation by interacting with PKM2 and regulating its phosphorylation status, thereby influence cellular metabolism and proliferation [14]. Moreover, RRM1 is associated with DNA damage induced by 5-FU and enhances the cytotoxic effect with cisplatin in treatment for EC [116]. Therefore, targeting RRM1 holds potential for enhancing the effectiveness of chemotherapeutic drugs in the treatment of EC.
Metabolic heterogeneity in EC stem cells
Esophageal Cancer Stem Cells (ECSCs) are a minority of cells in EC that possess self-renewal and tumor-forming capabilities. These cells are crucial in the initiation, proliferation, metastasis, and resistance to treatment in EC. Research shows that ECSCs adaptively rearrange their nutrient utilization in response to fluctuations in nutrient availability (Fig. 5). A trained stemness index model was used to evaluate EC patients, indicating that stemness characteristics are associated with poor prognosis and lower immune scores in EC. This highlights potential targets for CSC-mediated immunotherapy [117]. The study by Liu et al. shows that ESCC cell lines cultured under spheroid conditions show increased CSC phenotypes, including increased LDHA activity, chemotherapy resistance, and tumorigenic potential, all dependent on Hsp27 activation. Additionally, these cells undergo metabolic reprogramming characterized by heightened glycolytic and OXPHOS activities, governed by the Hsp27-AKT-HK2 pathway. This research highlights the critical role of the Hsp27-AKT-HK2 pathway in modulating the robust metabolic activity and stem cell properties of ECSCs [118]. CDK7 can phosphorylate nuclear Yes-associated protein 1 (YAP), enhancing the transcriptional activity and promoting the expression of D-lactate dehydrogenase (LDHD). Studies have demonstrated that LDHD expression is notably elevated in ECSCs and correlates with poor patient prognosis. By facilitating the clearance of D-lactate and production of pyruvate, the CDK7-YAP-LDHD axis enables ECSCs to evade ferroptosis and meet their high energy demands for self-renewal [119]. This study highlights the potential of the CDK7-YAP-LDHD axis in ESCC treatment and suggests that targeting the metabolic checkpoint could be an effective strategy for treating EC patients. Under hypoxic conditions, glycolysis and stemness of EC cells are enhanced. Mechanism analyses uncovered that hypoxia induces lactylation at the K147 site of Axin1, promoting its ubiquitination and degradation, thereby enhancing the stemness of EC cells by upregulating LDHA expression [120]. Another mechanistic study demonstrated that, under hypoxia, EC cells can enhance the expression of SHMT2, which in turn regulates the protein levels of GLUT1 and HK2, leading to increased glycolysis and stemness in EC cells [88]. These studies collectively demonstrate the existence of multiple mechanisms regulating stemness in EC, suggesting that inhibiting the stemness of EC cells represents a feasible strategy for EC treatment. Future research should focus on uncovering the key regulatory mechanisms that enhance EC stemness, as well as the specific role of EC stem cells in tumor progression, thereby providing new therapeutic approaches for targeting EC stem cells to suppress EC.

ECSC exhibits stronger glycolytic and oxidative phosphorylation activities. The YAP signaling pathway contributes to enhanced tumorigenic potential, while the AKT/mTOR signaling pathway increases drug resistance in ECSC. Enzymes are highlighted in red, whereas regulators of key enzymes are depicted in green. Figure created with BioRender.com.
Target-specific metabolic enzyme
In developing treatment strategies for EC, focusing on metabolic enzymes is crucial. EC cells frequently reprogram their metabolic networks to support rapid proliferation and adapt to the microenvironment, exhibiting significant alterations in glucose, amino acid, and nucleotide metabolism. These metabolic adaptations not only facilitate tumor growth and dissemination but also present novel therapeutic opportunities. Understanding how these metabolic enzymes function under pathological conditions lays a solid foundation for designing targeted treatments. By strategically inhibiting these critical enzymes, we can disrupt the survival mechanisms of tumor cells, thereby curbing their growth and invasive potential. This section will examine current therapeutic approaches targeting metabolic enzymes in EC, with a focus on various inhibitors that are either in clinical use or under investigation (Table 1), with the goal of providing more effective and safer treatment options.
Targeting glucose metabolism
GLUT1 is considered a potential therapeutic target due to its high expression in various cancers. Currently, several inhibitors that specifically target and inhibit GLUT1 activity have been identified. These include natural products such as cytochalasin B and quercetin, as well as synthetic compounds like WZB117, Fasentin, and BAY-876. BAY-876 is a potent GLUT1 inhibitor identified in a screen of ~3 million compounds [121]. Research has shown that BAY-876 significantly promotes apoptosis in ESCC cells and enhances the response of ESCC to cisplatin treatment. This suggests that GLUT1 inhibition may provide a potential novel therapeutic approach and BAY-876 could serve as a promising candidate drug for the treatment of ESCC [122]. Although BAY-876 demonstrates significant selective inhibition of GLUT1 and exhibits notable antitumor activity against EC in in vitro experiments and in vivo animal models, its safety and efficacy in humans remains uncertain. Significant progress has been made in the functional research of HK2 in EC, and several mature inhibitors have been developed, including 2-deoxy-D-glucose (2-DG), Benserazide (Benz), and Lonidamine. Among these, 2-DG is a glucose analog that competitively inhibits HK2 function to block the glycolytic pathway. Experimental data suggest that 2-DG inhibit the formation of ESCC spheroids and cell proliferation, reduce the stemness of cancer cells, and decrease tumor incidence [118]. Moreover, 2-DG has shown promise as a radiotherapy sensitizer in Phase I/II clinical trials, making it a potential therapeutic candidate for EC. In addition to 2-DG, the antifungal drug Sulconazole has been reported to disrupt glycolysis and induce PANoptosis in EC cells by inhibiting HKs. The new finding provides strong laboratory evidence supporting the potential clinical application of Sulconazole in EC treatment [123].
PFKs is a key enzyme regulating glycolysis. Investigation has revealed that the antipsychotic drug Trifluoperazine can inhibit PFKL, a subtype of PFK, suppressing the growth of ESCC both in vitro and in vivo [124]. PFKFB3 is a core protein that regulates glycolytic metabolism, and its expression levels and post-translational modifications are crucial for cancer progression. Key inhibitors of PFKFB3 include 3PO, PFK15, and PFK158, with PFK158 having advanced to clinical trials [125]. In ESCC, findings confirm that PFK15 can inhibit tumor growth in both cellular models and nude mouse xenograft models. Meanwhile, ESCC cells increased the phosphorylation of PFKFB3 at serine 461 under metabolic stress, promoting its translocation to the nucleus to regulate the expression of genes associated with immune evasion. The combination of PFK15 with PD-1 monoclonal antibody may enhance therapeutic efficacy, providing a novel combination treatment strategy for ESCC [126].
The high expression of PKM2 in tumor cells is very common, and inhibitors targeting PKM2 have attracted significant attention in cancer research. PKM2 inhibitors, such as Shikonin, TEPP-46, ML-265, and BAY-1436032 have demonstrated significant potential in inhibiting tumor cell glycolysis and metabolic reprogramming. For instance, the natural compound Shikonin suppresses aerobic glycolysis in ESCC cells by inhibiting phosphorylated PKM2, effectively reducing ESCC growth both in vitro and in vivo, making it a valuable therapeutic option for ESCC [127]. In addition, Dihydroartemisinin, a clinically used antimalarial agent, has been reported to exert anti-ESCC effects by inhibiting PKM2 [128]. LDHA is highly expressed in tumor cells, supporting their metabolic demands and sustaining the Warburg effect. Several specific inhibitors, such as FX11, GNE-140, and Galloflavin, have shown potential for anti-tumor applications by targeting LDHA in preclinical studies. Sodium oxamate (SO), a pyruvate analog, is one of the earliest identified inhibitors of LDHA. In a TNF-α induced EC cells migration model, SO was found to effectively inhibit tumor cell migration by reducing lactate production through the suppression of LDHA levels. This suggests that LDHA inhibitors, which restrict cancer cell migration driven by inflammatory processes, could be considered as an adjunct to standard therapy for EC patients [48]. Dichloroacetic acid (DCA) is a small molecule compound that has been found in metabolic studies to inhibit PDK phosphorylation and reverse the “Warburg effect” of tumor cells, eventually resulting in the induction of mitochondria-driven apoptosis [129]. Experimental data suggest that combining DCA with the hyaluronic acid synthesis inhibitor 4-methylumbelliferone can significantly enhance the anti-EC effect [130]. CPI-613, a derivative of the PDH complex cofactor lipoate, disrupts normal mitochondrial function by inhibiting PDH activity. Originally developed as a first-in-class drug targeting AML, CPI-613 has been shown to suppress the stemness of ESCC cells by inhibiting PDH activity. This research highlights the potential of CPI-613 in the treatment of ESCC [131].
IDH1 and IDH2 are key enzymes in the TCA cycle, and mutations in these enzymes are frequently found in many cancer cells. The most common mutations are the R132 mutation in IDH1 and the R140/R172 mutations in IDH2. Drugs developed to target cancers harboring IDH1 or IDH2 mutations, such as AML and gliomas, including Ivosidenib (AG-120), Enasidenib (AG-221), and Vorasidenib (AG-881), have demonstrated promising therapeutic effects in clinical applications. As previously mentioned, while the expression of IDHs is significantly elevated in EC, the low frequency of IDHs mutations has limited the use of IDHs inhibitors as a primary therapeutic strategy. With advancements in molecular targeted therapy, if IDH mutations are identified in specific subtypes of EC, IDHs inhibitors such as AG-120 and AG-221 may emerge as a potential therapeutic option [132]. G6PD is a key enzyme in the PPP, and its activity is closely associated with tumor cell proliferation and oxidative stress resistance, making it a potential anticancer target. Currently, small molecules such as DHEA, 6-AN, and the natural product Polydatin are commonly used as inhibitors of G6PD. Research has demonstrated that treatment of ESCC cells with Polydatin significantly inhibits G6PD activity and reduces intracellular NADPH levels, thereby altering the metabolic state of the cells and suppressing ESCC cell proliferation [63]. Consequently, Polydatin may represent a promising compound for adjuvant therapy in EC.
Targeting amino acid metabolism
Tumor cells typically depend on specific amino acids to support their proliferation and survival, making the regulation of amino acid supply and metabolic pathways an effective strategy for inhibiting tumor growth. As a key enzyme in glutamine metabolism, inhibiting GLS activity has been proven to be an effective anti-tumor strategy. The primary GLS inhibitors currently available include CB-839, and 6-Diazo-5-oxo-L-norleucine (DON). In EC research, CB-839 has demonstrated the ability to directly inhibit GLS activity and decrease its expression, disrupt the interaction between GLS and PDK1, and suppress both glycolysis and glutamine metabolism in tumor cells, thereby exerting a significant inhibitory effect on EC [13]. Furthermore, investigations have revealed that CB-839 exerts a more pronounced inhibitory effect on ESCC cells with dysregulation of the Fbxo4-cyclin D1 axis. When combined with mitochondrial respiration inhibitors, such as Metformin or Phenformin, CB-839 can produce a synergistic effect. This dysregulated Fbxo4-cyclin D1 axis promotes the resistance of ESCC cells to CDK4/6 inhibitors. Introducing CB-839, which targets the metabolic vulnerabilities of tumor cells, enhances tumor cell sensitivity to treatment, offering a novel potential strategy to overcome ESCC resistance [77]. Therefore, CB-839 may be an effective drug for combination therapy in ESCC.
IDO1 and TDO are key enzymes that regulate tryptophan metabolism and are crucial in tumor immune modulation. Currently, several IDO1 inhibitors, including Epacadostat, Indoximod, and Navoximod, have entered clinical trial stages. These inhibitors aim to enhance PD-L1-mediated antitumor immune responses by blocking the tryptophan metabolic pathway. In EC, the clinical trial NCT03196232 evaluated the efficacy of the IDO1 inhibitor Epacadostat in combination with Pembrolizumab in patients with metastatic or unresectable gastroesophageal junction and gastric adenocarcinoma. However, the clinical trial did not achieve the anticipated primary endpoint. The combination of Epacadostat and Pembrolizumab did not significantly improve progression-free survival or overall response rate compared to Pembrolizumab alone [133]. The failure of Epacadostat in clinical trials may be attributed to its insufficient ability to effectively inhibit IDO1 expression at the tumor site. To address this challenge, researchers have developed nanosheet carriers to deliver IDO1 inhibitors directly to the tumor, thereby enhancing the antitumor efficacy of chimeric antigen receptor T cells (CAR-T) in ESCC [134]. Despite the controversy surrounding the efficacy of IDO1 inhibitors in cancer immunotherapy, researchers remain optimistic that their effectiveness may be enhanced in specific subtypes of EC patients or through combination with other therapies. Compared to IDO1, the development of TDO inhibitors has progressed relatively slowly, and there are currently no TDO inhibitors widely applied in clinical treatment. However, recent studies indicate that compound 680C91 can inhibit the proliferation of ESCC cells and tumor growth by inhibiting TDO2, preventing the polarization of M2 macrophages, and thereby enhancing the tumor immune response. 680C91 is a promising TDO2 inhibitor that may serve as an effective molecule for targeted therapy in ESCC [84].
Targeting the inhibition of arginine metabolism represents a promising anticancer strategy, particularly in the context of immunosuppressive tumor microenvironments. ODC is the rate-limiting enzyme in the polyamine synthesis pathway of arginine metabolism. Research findings reveal that the irreversible inhibitor of ODC, Difluoromethylornithine (DFMO), can inhibit the production of mucosal polyamines and promote the expression of genes associated with chemoprevention, such as RPL11, PLXB1, and TAF2. This suggests that DFMO may be utilized for the chemoprevention of EAC in patients with Barrett’s esophagus (BE) [135]. Arginase is highly expressed in EC. Evidence suggests that CB-1158 increased tumor-infiltrating CD8+ T cells and NK cells by inhibiting the activity of arginase1. Additionally, CB-1158 upregulates the expression of inflammatory cytokines and interferon-induced genes, shifting the tumor microenvironment towards a pro-inflammatory state. This mechanism effectively suppresses tumor growth in various mouse tumor models, including EC [136]. iNOS inhibitors have shown broad application potential in various diseases, including tumors, inflammation, neurological disorders, and cardiovascular diseases. A study found that the iNOS inhibitor PBIT significantly suppresses the occurrence and progression of esophageal tumors by reducing nitric oxide production in NMBA-induced tumorigenesis in the rat esophagus, suggesting its potential as a chemopreventive agent for EC [137]. Therefore, in the anti-EC strategies targeting arginine metabolism, ODC, arginase, and iNOS may serve as effective therapeutic targets. The inhibitors DFMO, CB-1158, and PBIT exhibit potential in suppressing the progression of EC and warrant further investigation.
Solute carrier family is a crucial group of proteins located on cell membranes, responsible for the transport of various nutrients, metabolites, and drugs. These proteins play a pivotal role in regulating tumor metabolism. Targeting specific members of the SLC family to block the absorption and metabolic pathways of nutrients in tumor cells can effectively inhibit tumor growth and dissemination. LAT1, also known as SLC7A5, is a key protein in the SLC family responsible for transporting most neutral amino acids. Frequently used LAT1 inhibitors include JPH203, BCH, and KMH-233. Studies have demonstrated that the LAT1 inhibitor BCH can significantly suppress the growth of ESCC both in vitro and in vivo by inhibiting mTOR signaling and cell cycle progression [138]. Another LAT1 inhibitor, JPH203, has shown anticancer potential in clinical trials for pancreatic and colorectal cancers. However, studies indicate that JPH203 exhibits weaker inhibitory activity in EC [139]. SLC7A11 is another member of the SLC family linked to tumor metabolism and is significantly upregulated in EC. This protein primarily facilitates the exchange of extracellular glutamate and cysteine, playing a vital role in GSH synthesis, thereby helping maintain the cell’s antioxidant capacity. Inhibitors of SLC7A11, such as Erastin and Sulfasalazine, have been extensively studied in various cancer, particularly in tumors that develop resistance to conventional therapies, as they can enhance therapeutic efficacy by inducing ferroptosis. Research has found that Erastin can significantly inhibit the function of SLC7A11, alleviating the malignant phenotype of ESCC cells and downregulating key ferroptosis-related molecules GPX4 and DHODH [140]. Moreover, Erastin significantly inhibited the cell viability of ECSCs, and the inhibitory effects became more pronounced with increasing drug concentration. Compared to conventional cancer cells, Erastin demonstrated a stronger inhibitory effect on ECSCs [141]. SLC1A5, also known as ASCT2, plays a key role in neutral amino acid transport, particularly glutamine. GPNA has been reported to inhibit the uptake of glutamine in EAC cells by targeting and inhibiting SLC1A5, thereby suppressing tumor cells proliferation [142]. Therefore, inhibitors of the solute carrier family, such as BCH, Erastin, and GPNA, may be potential candidate molecules for the treatment of EC.
Glutaminyl cyclase (QC) is an enzyme that catalyzes the conversion of glutamine residues into pyroglutamate, a process involved in key mechanisms of tumor immune evasion. Consequently, QC inhibitors have emerged as potential immune therapeutic targets. Mechanistic studies demonstrated that the small molecule SEN177 significantly enhances macrophage- and neutrophil-mediated antibody-dependent cellular phagocytosis (ADCP) and antibody-dependent cell-mediated cytotoxicity (ADCC) by targeting and inhibiting QC, thereby improving the therapeutic efficacy of EGFR antibodies against EC [143]. Therefore, the small molecule SEN177 may serve as an adjuvant in antibody therapies for EC.
Targeting nucleotide metabolism
Targeting nucleotide metabolism is a common clinical strategy in cancer treatment. Drugs that inhibit the synthesis of DNA or RNA by interfering with cellular nucleotide metabolic pathways are referred to as antimetabolites. Since the mid-20th century, antimetabolites have been a mainstay of chemotherapy, playing a critical role particularly in the treatment of hematological malignancies and various solid tumors. 5-FU is a commonly used chemotherapeutic agent that inhibits TS to block DNA synthesis, thereby suppressing tumor cell proliferation. However, clinical data confirm that the therapeutic efficacy of 5-FU in advanced EC is limited, possibly as a result of various resistance mechanisms, including overexpression of TS in EC cells, alterations in enzymes related to 5-FU metabolism, and enhanced drug efflux mechanisms [144, 145]. Methotrexate (MTX), a widely used antimetabolite that targets dihydrofolate reductase (DHFR), has shown notable therapeutic efficacy in treating EC. However, its clinical use is constrained by adverse effects, such as liver and kidney toxicity, as well as immunosuppression [146]. Several drugs are clinically available that can target and inhibit RNR, including Gemcitabine, Hydroxyurea, and Clofarabine. Gemcitabine has been shown to effectively control the progression of ESCC in clinical applications, significantly prolonging patient survival. However, Gemcitabine is associated with side effects such as myelosuppression, nausea, and alopecia, which limits its application [147]. Additionally, studies have revealed that Osalmid can enhance the DNA damage response in ESCC cells by inhibiting the activity of the RNR small subunit RRM2, significantly increasing the radiosensitivity of tumor cells [148]. Several studies have explored the potential of Metformin in the treatment of ESCC. It has been found that Metformin can alter nucleotide metabolism in EC cells, leading to an increase in intracellular dTTP pools and a dilution of toxic 5-FU anabolites, which weakens the therapeutic efficacy of 5-FU. However, Metformin has also been shown to increase the expression of deoxycytidine kinase (DCK), thereby enhancing the sensitivity of tumor cells to Gemcitabine. This finding indicates that a combination therapy of Metformin and Gemcitabine may represent a potential new strategy for treating ESCC [149]. In summary, although several drugs have demonstrated significant efficacy in treating EC by targeting nucleotide metabolism, the pronounced side effects from inhibiting normal cells cannot be overlooked. Innovative therapeutic approaches, such as combination therapies, may help better address the treatment challenges posed by nucleotide metabolism dysregulation associated with EC.
Perspective
The metabolic characteristics of tumors exhibit significant heterogeneity across different types of cancer. Furthermore, there are notable metabolic differences within the same tumor at different stages of development and in various regions of the tumor. This heterogeneity primarily arises from complex factors both inside and outside the tumor cells, such as the tumor microenvironment and genetic factors within the tumor cells, as well as the interactions between these two elements. EC is a highly heterogeneous malignancy, its metabolic characteristics showing significant variations across different types and stages. In ESCC, tumor cells typically rely on the reprogramming of glucose metabolism, exhibiting elevated levels of glycolysis. Even under aerobic conditions, these cells preferentially engage glycolytic pathways to meet the energy demands of rapid proliferation. In contrast, EAC tends to utilize fatty acid oxidation as a primary energy source, and may also display a dependency on glutamine metabolism, which provides carbon and nitrogen sources necessary for the synthesis of nucleic acids and proteins in rapidly dividing cancer cells [150]. The metabolic characteristics of EC differ across various stages of disease progression. In the early stages of EC, cancer cell metabolism may still closely resemble normal cell metabolism, with relatively mild changes in metabolic pathways. However, the balance between glycolysis and oxidative phosphorylation begins to shift. As the tumor advances, particularly in late-stage and metastatic EC, the metabolic profile of cancer cells undergoes significant alterations, exhibiting a greater dependence on glycolysis or active regulation of other metabolic pathways. The metabolic heterogeneity of EC contributes to its varying responses and resistance to chemotherapy, radiotherapy, and targeted therapies. Therefore, personalized treatment approaches based on metabolic characteristics may offer a promising strategy for improving therapeutic outcomes in EC.
This review summarizes the key enzyme changes associated with glucose, amino acid, and nucleotide metabolism during the progression of EC and highlights several promising therapeutic agents. These agents may provide more precise strategies for targeting metabolic reprogramming in EC treatment (Fig. 6). For example, with selective inhibition of key pathways of glycolysis or glutamine metabolism, these drugs can effectively weaken the energy supply of cancer cells. By targeting key enzymes involved in nucleotide metabolism, such as MTH1 and RNR, effectively disrupts DNA synthesis and repair in tumor cells. Combining inhibitors of these key metabolic enzymes with existing EC therapies may yield potential synergistic effects. In addition, there are other approaches to control the progression of EC by regulating its metabolic reprogramming. For instance, dietary adjustments that enhance healthy energy supply while minimizing external metabolic disruptions are important measures to control metabolic imbalances in EC. Furthermore, personalized treatment strategies for EC, including gene editing and immunometabolic therapies, directly target the metabolic pathways of tumor cells and inhibit tumor growth and metastasis by altering the tumor microenvironment. In the treatment of EC, developing multi-target combination therapies and precision medicine could maximize therapeutic efficacy while minimizing side effects by integrating multiple treatment modalities. These emerging therapeutic approaches are expected to provide more effective options for EC patients. Thus, targeting metabolic reprogramming in EC represents a key area of research in EC treatment, offering new insights and approaches for clinical treatment.

The figure illustrates the anatomical segmentation of the esophagus, diagnostic methods including endoscopy, imaging, and tumor marker examination, and targeted treatment strategies focusing on glucose, nucleotide, and amino acid metabolism. Future treatment strategies encompass multi-target combination therapy, precision medicine, and interactive tumor immune metabolism therapy, addressing the challenges and prospects in EC management. Figure created with BioRender.com.
Responses