Extravascular coagulation regulates haemostasis independently of activated platelet surfaces in an in vivo mouse model
Introduction
When bleeding occurs, a haemostatic plug forms to prevent blood loss. Traditionally, it is believed that platelet activation and coagulation occur simultaneously at the injury site, relying on phosphatidylserine (PS) on activated platelets for effective clot formation1,2,3,4. However, this view does not explain why platelet dysfunction rarely results in severe bleeding, whereas coagulation disorders often do, especially in deep tissues5.
The ancestors of platelets are believed to be amebocytes found in horseshoe crab haemolymph6. These cells release serine proteases, such as prochelicerase C and prochelicerase B7, which are thought to be the ancestors of coagulation factors. These proteases initiate a cascade of reactions for wound healing8. Furthermore, activated amebocytes release serine proteases, antimicrobial peptides, and lectins9, but unlike human platelets, the cells themselves cannot seal wounds or initiate cascade reactions on their surface10,11. Given that coagulation factors are evolutionarily more primitive than platelets are, we hypothesize that coagulation, rather than platelets, is the trigger for haemostasis.
Recent advancements in in vivo imaging techniques have been utilized to examine the process of thrombus formation12,13,14,15. Two common in vivo imaging techniques for observing thrombus formation are as follows: (1) microscopy after oxidative endothelial injury from chemicals such as ferric chloride16,17,18 and (2) microscopy following laser-induced endothelial injury12,13,15,19,20,21,22,23. In the oxidative injury model, vascular endothelial desquamation is typically observed after chemical stimulation, although some reports note intact vascular endothelium24. Conversely, the conventional laser injury model maintains an intact vascular endothelium, with tissue factor release at the laser site driving thrombus formation12,19. Both methods lack bleeding. The presence of a thrombus without bleeding is deemed pathological, leading previous in vivo imaging methods to focus on assessing pathological thromboses rather than haemostasis. In contrast, Bergmeier et al. described a method for inducing external bleeding by applying penetrating laser injury to exposed blood vessels, followed by observation of haemostasis25,26. However, to date, no methods have been reported for confirming haemostasis following internal bleeding, which frequently occurs in everyday life outside traumatic events. In this study, we developed an in vivo imaging method to observe the early haemostatic response after internal bleeding by disrupting the vascular endothelium via two-photon excitation. Our method confirmed that the haemostatic response remained unaffected in mice with impaired platelet surface coagulation. In contrast, a delayed haemostatic response was observed in mice with low tissue factor expression. These findings indicate that extravascular coagulation acts as the primary trigger for haemostasis during internal bleeding, whereas coagulation on the activated platelet surface is not essential for this process. These novel findings clarify the discrepancy between the traditional understanding of haemostasis and clinical observations regarding platelet dysfunction. These findings could also contribute to improved management of haemostatic abnormalities and to advances in antithrombotic therapy.
Results
Fibrin is formed outside blood vessels
We developed an in vivo imaging method to observe bleeding and haemostatic plug formation in veins and arteries, with a focus on platelets and coagulation after internal bleeding. In C57BL/6J (wild-type) mice, we observed that venous and arterial haemostatic plugs contained platelet aggregates inside the vessel, fibrin in platelet-free areas outside the vessel, and coexisting platelet and fibrin clots at the boundaries (Fig. 1a, b).

Representative images of haemostatic plug formation after venous (a) and arterial (b) internal bleeding. The mice were intravenously injected with dextran-rhodamine B, Alexa Fluor 488-conjugated fibrinogen and the platelet imaging antibody X649 prior to observation. After two-photon excitation injury (circle of dots), platelet aggregate formation in the lumen (magenta triangle) and extravascular plasma leakage (red triangle) were observed. After arterial internal bleeding, red blood cell leakage was observed (white asterisk). Fibrin is formed in the extravascular area (green triangle). The haemostatic plug consisted of a platelet aggregate in the lumen, extravascular fibrin, and platelets and fibrin at the boundary of the lumen and extravascular space (white triangle). Scale bar = 20 μm. Green: fibrin/fibrinogen. Magenta: platelets. Red: plasma.
Haemostasis after arterial internal bleeding was completed by cessation of plasma leakage following cessation of blood cell leakage
In our experiments, venous internal bleeding included plasma leakage, identified by fluorescent dextran leakage, with minimal blood cell leakage. In contrast, arterial internal bleeding resulted in both blood cell and plasma leakage, with red blood cells visible as black silhouettes (Fig. 1a, b). To confirm how blood cell and plasma leakage ceases during haemostasis after arterial internal bleeding, we analysed images of significant vascular damage (25–40 mm) that allowed clear observation of blood cell leakage. Consequently, haemostasis was achieved first with the cessation of blood cell leakage, followed by the cessation of plasma leakage. Low plasma leakage results in slight fluctuations in the dextran signal, making it difficult to determine when leakage stops. However, during blood cell leakage, clear extravascular leakage of fluorescent dextran was observed in all mice. Although blood cell leakage recurred, it ceased when platelets clog the vascular injury site (Fig. 2).

Representative images of haemostatic plug formation after arterial internal bleeding from a large injury. The mice were intravenously injected with dextran-rhodamine B, Alexa Fluor 488-conjugated fibrinogen, and the platelet imaging antibody X649 prior to observation. After two-photon excitation injury (circle of dots), extravascular plasma leakage (red triangle) and red blood cell leakage were observed (white asterisk). Vasoconstriction temporarily reduced blood leakage, but it resumed when vasoconstriction was released. Fibrin begins to form in slow-flowing areas of the extravascular space (green triangle). Exacerbated blood cell leakage stopped when platelets clogged the site of vessel rupture (magenta triangle). Fibrin formation proceeded in the area of slowed flow caused by platelet blockage, resulting in the cessation of plasma leakage. Scale bar = 20 μm. Green: fibrin/fibrinogen. Magenta: platelets. Red: plasma.
To investigate platelet entrapment outside the vessel, we observed bleeding sites in wild-type mice via two-photon imaging, which is known for its ability to visualize collagen27. We observed that leaking platelets were attached to fibrin where collagen was disrupted. Similar findings were noted in platelet-specific anoctamin-6 (TMEM16F) knockout (Ano6 cKO) mice, a murine model of Scott’s syndrome characterized by impaired expression of PS on platelets28. This finding indicated that fibrin entrapped leaked platelets independently of platelet activation (Fig. 3).

Representative images of haemostatic plugs after large arterial injury. The mice were intravenously injected with dextran-rhodamine B, Alexa Fluor 488-conjugated fibrinogen, and the platelet imaging antibody X649 prior to observation. Vessel observation before the arterial injury and after haemostatic plug formation via two-photon imaging revealed collagen (blue triangle) and platelets attached to the fibrin network (green triangle), where collagen ruptured. Similar findings were observed in Ano6 cKO mice with impaired platelet coagulation. Scale bar = 20 μm. In the confocal images, green indicates fibrin/fibrinogen, magenta indicates platelets, and red indicates plasma. In two-photon images, yellow-green indicates fibrin/fibrinogen, red indicates plasma, and blue indicates collagen.
Irreversible platelet activation was not observed in the early phase of haemostatic plug formation
We then investigated the activation status of platelets in haemostasis by examining CD62P, a marker of irreversibly activated platelets. While CD62P was detected in platelets that clog the liver vasculature after intravenous thrombin injection (positive control, Fig. 4a), it was not detected in the in vivo bleeding/haemostasis model, in accordance with our hypothesis (Fig. 4b). These findings suggest that platelets exhibit limited activation during the early phase of haemostatic plug formation, with coagulation predominantly occurring outside the vessel.
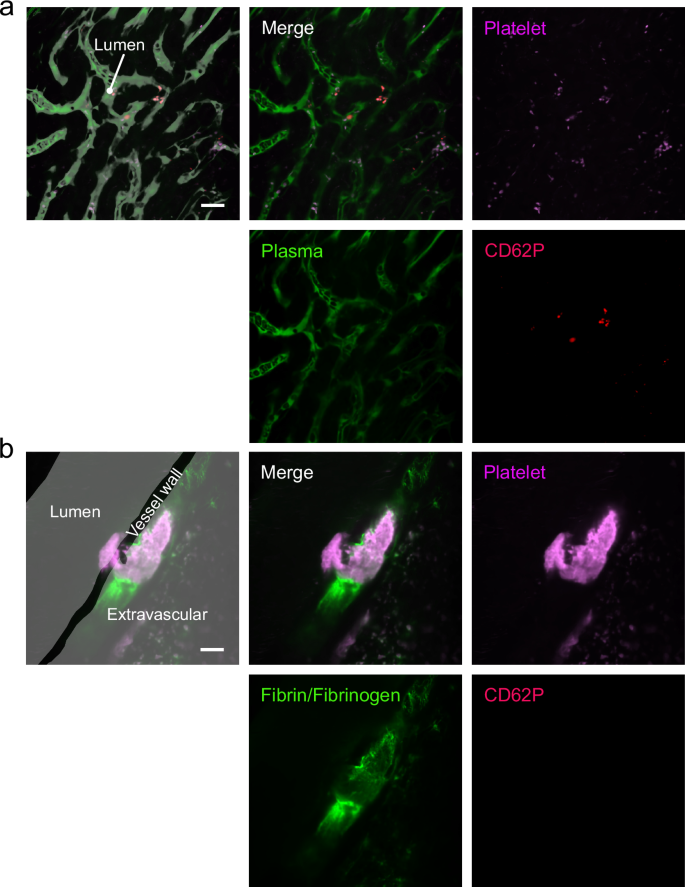
Representative images of platelets that clog the liver vasculature after systemic thrombin injection (a) and haemostatic plugs that formed after large arterial injury (b). For arterial haemostatic plug observation, the mice were injected with Alexa Fluor 488-conjugated fibrinogen, PE-conjugated rat anti-mouse CD62P antibody, and platelet imaging antibody X649 prior to observation. For platelet observation after thrombin injection, the mice were injected with FITC-dextran, PE-conjugated rat anti-mouse CD62P antibody, and X649 antibody prior to observation. CD62P expression was detected on platelets that clog the liver vasculature (a) but was not detected in the in vivo internal bleeding/haemostasis model (b). Scale bar = 20 μm. Green: fibrin/fibrinogen. Magenta: platelets. Red: plasma.
Quantitative analysis of haemostatic plug formation in wild-type, Ano6 cKO, platelet-depleted, FVIII KO, and low-tissue-factor mice
To investigate the role of platelets and coagulation in haemostatic plug formation, we analysed images from wild-type, platelet-depleted (administered antibodies for platelet depletion with platelet counts of approximately 3% of the predose level), and three genetically modified mouse strains, Ano6 cKO; coagulation factor VIII-deficient (FVIII KO), which represent severe haemophilia A; and low-tissue-factor-expressing (L-TF) mice, which are indicative of an impaired extrinsic coagulation pathway, were used. We focused on lesions within specific vascular injury ranges—25–40 μm for veins (wild-type, n = 6 lesions of 5 animals; Ano6 cKO, n = 5 lesions of 5 animals; platelet-depleted, n = 5 lesions of 5 animals; FVIII KO, n = 6 lesions of 4 animals; and L-TF mice, n = 16 lesions of 4 animals) and 10–25 μm for arteries (wild-type, n = 6 lesions of 6 animals; Ano6 cKO, n = 7 lesions of 6 animals; platelet-depleted, n = 5 lesions of 5 animals; FVIII KO, n = 5 lesions of 5 animals; and L-TF mice, n = 6 lesions of 6 animals)—ensuring accuracy in our evaluations. Representative images and movies are shown in Figs. 5, 6, and Supplementary Movies 1–10. Figures 7a, 8a, Supplementary Figs. 1 and 2 show time-series data obtained from individual mice, while Figs. 7b–f and 8b–h present extracted data related to platelet aggregates, fibrin, and bleeding.

Representative images of haemostatic plug formation after venous internal bleeding in wild-type, Ano6 cKO, platelet-depleted, FVIII KO, and L-TF mice. The mice were intravenously injected with dextran-rhodamine B, Alexa Fluor 488-conjugated fibrinogen, and the platelet imaging antibody X649 prior to observation. Scale bar = 20 μm. Green: fibrin/fibrinogen. Magenta: platelets. Red: plasma.

Representative images of haemostatic plug formation after arterial internal bleeding in wild-type, Ano6 cKO, platelet-depleted, FVIII KO, and L-TF mice. The mice were intravenously injected with dextran-rhodamine B, Alexa Fluor 488-conjugated fibrinogen, and the platelet imaging antibody X649 prior to observation. Scale bar = 20 μm. Green: fibrin/fibrinogen. Magenta: platelets. Red: plasma.

Measurements of platelet aggregates, fibrin, and plasma leakage were taken from experimental mice (a) (wild-type, n = 6 lesions from 5 animals; Ano6 cKO, n = 5 lesions from 5 animals; platelet-depleted, n = 5 lesions from 5 animals; FVIII KO, n = 6 lesions from 4 animals; and L-TF mice, n = 16 lesions from 4 animals). Platelet aggregates were assessed by height from the vascular endothelium. Fibrin was quantified as the ratio of the fibrin area over time, normalized to the end of the observation period. Plasma leakage was expressed relative to the maximum signal outside the vessel. Box-and-whisker plot of the peak platelet aggregate height (b). Peak platelet aggregate height at 0–120 and 120–300 s (c). The increase in the fibrin area accelerated, as indicated by the ratio (d). Time required for the fibrin area to reach half the area at the end of observation (e). Area under the curve (AUC) of plasma leakage (f). *<0.05, **<0.01. c Wilcoxon signed-rank test; b, d–f Steel–Dwass test and Monte Carlo simulation.

Measurements of platelet aggregates, fibrin, and plasma leakage were taken from experimental mice (a) (wild-type, n = 6 lesions of 6 animals; Ano6 cKO, n = 7 lesions of 6 animals; platelet-depleted, n = 5 lesions of 5 animals; FVIII KO, n = 5 lesions of 5 animals; and L-TF mice, n = 6 lesions of 6 animals). Platelet aggregates were assessed by height from the vascular endothelium. Fibrin was quantified as the ratio of the fibrin area over time, normalized to the end of the observation period. Plasma leakage was expressed relative to the maximum signal outside the vessel. Box-and-whisker plot of the peak platelet aggregate height (b). Peak platelet aggregate height at 0–120 and 120–300 s (c). The increase in the fibrin area accelerated, as indicated by the ratio (d). Time required for the fibrin area to reach half the area at the end of observation (e). Area under the curve (AUC) of plasma leakage (f). The time from the start of bleeding to the cessation of blood cell leakage (g). The extent of platelet aggregate extrusion from the vessel was evaluated by measuring the distance from the vessel wall to the platelets, as shown in (h). †<0.1, *<0.05, **<0.01. c Wilcoxon signed-rank test; b, d–h Steel–Dwass test and Monte Carlo simulation.
Extravascular coagulation is important for stable platelet aggregate formation
Statistical analysis of platelet aggregates formed after venous internal bleeding revealed a significantly lower peak height in L-TF mice than in wild-type (p = 0.0024), Ano6 cKO (p = 0.0005), and FVIII KO mice (p = 0.0012) (Fig. 7b). A comparison of aggregates formed between 0–120 s (early-stage) and between 120–300 s (late-stage) revealed a lower peak height in the late-stage for both FVIII KO (p = 0.031) and L-TF mice (p < 0.0001) (Fig. 7c). After arterial bleeding, peak heights did not differ among mouse types (Fig. 8b), except in platelet-depleted mice. When comparing early- and late-stage platelet aggregates, the peak height tended to be lower in the late-stage in FVIII KO and L-TF mice (p = 0.063) (Fig. 8c). These findings suggest the importance of extravascular coagulation in venous and arterial platelet aggregates.
Extravascular coagulation proceeds independently of the activated platelet surface
As bleeding progresses, the formation of extravascular fibrin increases, except in L-TF mice. Thus, to assess coagulation efficiency, we quantified fibrin formation acceleration rather than the total fibrin area. After venous internal bleeding, FVIII KO mice exhibited significantly lower acceleration compared to wild-type (p = 0.0025), Ano6 cKO (p = 0.044), and platelet-depleted mice (p = 0.020) (Fig. 7d). No significant difference was observed between wild-type and Ano6 cKO mice (p = 0.30). Following arterial internal bleeding, while the acceleration in platelet-depleted mice tended to be greater than that in FVIII KO mice (p = 0.098), no significant differences were found among the groups (Fig. 8d). These findings suggest that extravascular fibrin formation occurs independently of activated platelet surfaces in both veins and arteries.
Extravascular fibrin formation is important for the cessation of both blood cell and plasma leakage
Under our experimental conditions, minimal blood cell leakage occurred after venous injury. In terms of arterial internal bleeding, the time from bleeding onset to cessation of blood cell leakage was significantly longer in platelet-depleted and L-TF mice than in wild-type and Ano6 cKO mice (wild-type vs. platelet-depleted, p = 0.014; wild-type vs. L-TF, p = 0.0045; Ano6 cKO vs. platelet-depleted, p = 0.0064; and Ano6 cKO vs. L-TF, p = 0.0018) (Fig. 8g). Platelet extravasation revealed a significantly greater distance from the vascular endothelium to extravasated platelets in L-TF mice than in wild-type (p = 0.0054) and Ano6 cKO mice (p = 0.033) (Fig. 8h). Additionally, plasma leakage from internal venous bleeding was significantly greater in L-TF mice than in wild-type (p < 0.0001), FVIII KO (p = 0.0023), and Ano6 cKO mice (p = 0.0005) (Fig. 7f), whereas arterial internal bleeding was significantly greater in L-TF mice than in wild-type mice (p = 0.027), with a trend towards platelet-depleted mice (p = 0.064) (Fig. 8f). These findings highlight the importance of extravascular fibrin formation in achieving complete haemostasis.
Discussion
Haemostatic plug formation is traditionally thought to involve efficient coagulation on the surface of activated platelets1,2,3,4, suggesting that platelet dysfunction should lead to severe bleeding. However, in the clinic, platelet dysfunction disorders, such as thrombasthenia or Scott syndrome, cause minimal bleeding, whereas coagulation disorders, such as haemophilia, lead to severe joint or muscle bleeding5. The aim of this study was to elucidate the sites and roles of coagulation in haemostatic plug formation through in vivo imaging of internal bleeding. These findings suggest that extravascular coagulation, rather than coagulation on activated platelets, is crucial in initiating and regulating haemostasis following internal bleeding.
In vascular injury induced by conventional one-photon excitation, damage occurs not only in the targeted area but also in the superficial and deep surrounding regions. In contrast, our method of inflicting vascular injury with two-photon excitation can induce localized damage at the targeted depth without injuring the areas traversed by the laser. In this model, blood leaks from the vessel into the surrounding undamaged tissue. In the external bleeding model of Bergmeier et al., haemostatic plugs are observed exclusively within the intravascular space and at the boundary between intravascular and extravascular regions25,26. In contrast, in our internal bleeding model, haemostatic plugs are more widely distributed, extending from the intravascular area into the extravascular space. To determine whether the haemostatic plug observed in our model, which is composed of extravascular fibrin, intravascular platelet aggregate, and a clot consisting of both platelets and fibrin at the vascular boundary (Fig. 1a, b), is specific to our model, we punctured the mouse medial saphenous vein or artery with a 30 G needle and applied compression to halt bleeding. The resulting plug, containing extravascular fibrin and intravascular platelet aggregates, was similar to our model (Supplementary Fig. 3). Thus, we conclude that the plugs observed in our model resemble those resulting from vascular injury in mammals.
A clot of platelets and fibrin formed at the intra/extravascular boundary following the formation of extravascular fibrin, which was similarly observed in Ano6 cKO mice (Fig. 5). Extravascular fibrin formed even in the platelet-depleted and Ano6 cKO mice (Figs. 5 and 6). These findings indicate that coagulation on the surface of platelets plays a minor role in fibrin formation in our internal bleeding/haemostasis model. Notably, platelets involved in early haemostasis did not express CD62P, a marker of irreversibly activated platelets that serve as a site of coagulation (Fig. 4), which aligns with prior findings of no PS exposure on platelets even up to 30 min after bleeding29. In contrast, Bergmeier et al. reported that fibrin formed primarily on platelets with PS exposure in an external bleeding model26. This difference may stem from the fact that, in our internal bleeding model, blood leakage into an enclosed space rich in tissue factor allows coagulation to proceed even in the absence of activated platelets. Sufficient thrombin generation occurs not only on activated platelets but also on tissue factor-bearing cells30, and extravascular cells support the formation of a dense, fibrinolysis-resistant fibrin network31. Our findings and previous reports suggest that early after internal bleeding, platelet activation is limited, preventing coagulation within blood vessels while allowing rapid fibrin formation outside vessels. This mechanism may reduce the risk of pathological thrombosis and prevent exsanguination.
In our study, venous internal bleeding primarily caused plasma leakage with minimal blood cell leakage, whereas arterial internal bleeding involved leakage of both. During arterial internal bleeding, haemostasis occurred first with cessation of blood cell leakage, followed by cessation of plasma leakage (Figs. 1b, 2, 8a, g). L-TF and platelet-depleted mice had a significantly longer time to cease blood cell leakage (Fig. 8g). Although plasma leakage did not differ between wild-type and Ano6 cKO mice, it increased in L-TF mice with venous bleeding (Fig. 7f) and in L-TF and platelet-depleted mice with arterial bleeding (Fig. 8f). These findings highlight the critical role of extravascular coagulation in haemostasis in both veins and arteries, with platelets potentially contributing additional roles in arterial haemostasis other than PS exposure. While fibrin formation rates were not significantly different among the wild-type, Ano6 cKO, and platelet-depleted mice (Figs. 7d and 8d), the platelet-depleted mice spent more time reaching half the final fibrin amount during arterial internal bleeding (Fig. 8e), indicating that platelets help regulate blood flow at injury sites, promoting effective coagulation.
To elucidate the role of extravascular coagulation in platelet aggregate formation (Fig. 1a, b), we compared the heights of platelet aggregates among various mouse groups. In veins, L-TF mice presented significantly lower maximum platelet aggregate heights (Fig. 7b), emphasizing the role of extravascular coagulation through the TF-triggered extrinsic pathway. In the arteries, the maximum platelet aggregate height did not significantly differ among the wild-type, Ano6 cKO, FVIII KO, and L-TF mice (Fig. 8b). However, when comparing early-stage (0–120 s) and late-stage (120–300 s) platelet aggregates in both veins and arteries, FVIII KO and L-TF mice exhibited lower late-stage platelet aggregates (Figs. 7c and 8c). These findings suggest that in veins, platelet aggregate formation involves TF-triggered coagulation from the early-stage, whereas in arteries, coagulation becomes significant in the late-stage, with early activation driven by shear stress32,33. Platelets are regulated by both shear stress and extravascular coagulation. Extravascular thrombin activates platelets via PAR1 receptors34 (and PAR3 and PAR4 receptors in mice35), whereas thrombin and factor Xa activate the vascular endothelium via PAR1 and PAR236. This dual regulation may explain the reduced platelet aggregate height in mice with impaired extravascular coagulation. The mechanism by which extravascular coagulation influences haemostatic plug formation, including intravascular platelet aggregate formation, seems reasonable, as it helps prevent excessive thrombus formation once haemostasis is achieved.
Limitations
This study has several limitations. First, the vascular injury was observed under restricted conditions, limiting our understanding of how it affects haemostatic plug and thrombus formation in different vessels or different extents of damage. Second, during arterial internal bleeding, vasoconstriction causes discrepancies in the timing of bleeding exacerbation among mice, highlighting the need to understand its role in haemostatic plug formation. Third, we injected human fibrinogen conjugated with a fluorescent dye for in vivo observation, which means that we may have underestimated fibrin thrombi since we did not observe mouse fibrin clots. Finally, the anaesthetic urethane may influence the results by inducing sympathetic nervous system excitement, and its use poses challenges for replicating study conditions, as it is no longer recommended in animal experiments. The results of the evaluation of the effects of anaesthetic urethane compared with alternative anaesthetics (a combination of medetomidine, butorphanol and midazolam) are given in the Supplementary Information.
Conclusion
Our understanding of the impact of extravascular coagulation on haemostasis after internal bleeding is shown in Fig. 9. Our findings, together with those of previous reports25,26, suggest that the platelet surface is the site of coagulation in external bleeding and that the extravascular tissue is the site of coagulation in internal bleeding, which may explain the phenotype of platelet dysfunction, with frequent nasal and gum bleeding5 (external bleeding) and coagulation disorders with frequent deep tissue bleeding5 (internal bleeding). These insights could inform clinical treatment strategies, such as administering coagulation factors with plasma transfusion in severe trauma, aligning with findings from the PAMPer37 and COMBAT38 clinical trials that highlight benefits in emergency settings. While many clotting factor replacements and mimetics are designed for prolonged retention in the bloodstream, optimizing their concentration outside the vasculature could enable drug development that enhances haemostasis while reducing thrombosis risk. Additionally, if fibrinolytic drugs used for thrombosis treatment could be engineered to remain inactive outside the vascular system, it may become possible to manage thrombosis more safely.

When vascular continuity is lost, blood cells and plasma leak out of the vessels (a). Extravascular fibrin and platelets captured by fibrin inhibit blood cell and plasma leakage (b). Coagulation proceeds extravascularly, and the generated thrombin accelerates extravascular fibrin formation and activates platelets in the lumen (c). As extravascular fibrin grows, plasma leakage ceases, resulting in complete haemostasis (d).
In conclusion, we demonstrated the critical role of extravascular coagulation in the formation of haemostatic plugs following internal bleeding via in vivo imaging. These findings have the potential to enhance our understanding of bleeding symptoms in clinical settings and contribute to the development of more effective and safer therapeutic drugs. Furthermore, this research method proved to be a valuable tool for evaluating such drugs.
Methods
Mice
Male C57BL/6J (wild-type) mice, FVIII-deficient (FVIII KO) mice (B6;129S-F8tm1Kaz/J)39 and low-TF-expressing (L-TF) mice40, all with a matching genetic background, were used. These mice were aged 8–13 weeks and weighed 22–25 g. Platelet-specific anoctamin-6 (TMEM16F) knockout (Ano6 cKO) mice were generated by crossing anoctamin-6 flox/flox (B6.Cg-Ano6tm1.1Naga)41 and Pf4-iCre transgenic mice42. Wild-type mice received an intravenous injection of 2 mg/kg R300 antibody (Emfret Analytics GmbH & Co., KG, Würzburg, Germany)43 to obtain platelet-depleted mice. Wild-type mice were purchased from Japan SLC, Inc. (Shizuoka, Japan). FVIII KO and Pf4-iCre mice were obtained from The Jackson Laboratory (Maine, USA). Anoctamin-6 flox/flox mice were provided by Riken BRC (Ibaraki, Japan). L-TF mice were provided by Prof. Nigel Mackman (University of North Carolina at Chapel Hill). Mice obtained from other facilities were housed in the Institute of Experimental Animal Science at Nara Medical University for at least one week to allow for acclimatisation. The mice were kept under controlled conditions (20–26 °C, 40–60% humidity) with a 12-h light/dark cycle and had ad libitum access to food and water. Anaesthesia was induced via intraperitoneal injection of urethane (1.5 mg/kg). After observation, the mice were euthanized by cervical dislocation. The depth of anaesthesia was confirmed by the hindlimb retraction reflex and the abdominal skin pain reflex. All animal experiments were approved by the Institutional Animal Care and Use Committee and strictly followed the guidelines for animal experiments of Nara Medical University (Permit Nos. 13320, 13323, and 13387).
In vivo imaging of internal bleeding and haemostatic plug formation after two-photon excitation injury
An A1R-MP microscope (Nikon, Japan) was used for visual analysis of bleeding and haemostatic plug formation in the testicular artery and medial saphenous vein. After sufficient anaesthesia was achieved, two small incisions were made in the median abdomen and medial thigh to observe blood vessels on the surface of the testis and under the skin of the thigh. Mice were injected via the jugular vein with dextran-rhodamine B (MW 70 kDa) (Merck KGaA, Darmstadt, Germany) (40 μg/g body weight), Alexa Fluor 488-conjugated fibrinogen (Thermo Fisher, Massachusetts, USA) (30 μg/g body weight) and X649 antibody (Emfret) (0.1 μg/g body weight), which labels platelets in mice without affecting their function. The mice were placed on a heated stage (Tokaihit, Shizuoka, Japan), and transverse sections of the testicular arteries and medial saphenous veins were visualized via confocal imaging (lens: Nikon Apo LWD ×40/1.15 WI λS, scanner zoom: 1.5×). The endothelium was cauterized by two-photon excitation with a pulsed laser at 800 nm (laser power: ~2064 mW) (laser source: COHERENT Verdi 18 W) (Coherent, Pennsylvania, U.S.A.), with an injury time of 0.8–1.6 s. Laser irradiation was stopped immediately after bleeding was obtained, and the irradiation area was 40 μm in diameter. After vascular injury, extravascular collagen was observed via two-photon microscopy at 930 nm in the same area. The total observation time was 5 min.
In vivo imaging of platelet activation
After the appropriate depth of anaesthesia was achieved, small incisions were made to observe the testis, and Alexa Fluor 488-conjugated fibrinogen, 25 μL of PE-conjugated rat anti-mouse CD62P antibody (Emfret), and X649 antibody were injected intravenously. Platelets involved in haemostasis were then observed in the testicular artery of wild-type mice via microscopy following two-photon excitation injury. In the positive control, small incisions were made in the median upper abdomen to observe the liver after it reached the appropriate depth of anaesthesia. FITC-dextran (MW 70 kDa) (Merck KGaA) (8 μg per g body weight), PE-conjugated rat anti-mouse CD62P antibody, X649 antibody, and 1 U per body thrombin (Fuji Pharma Co., Ltd., Tokyo, Japan) were injected intravenously, and platelets were observed in the liver vasculature via microscopy.
Image analysis
For quantitative analysis of bleeding and fibrin/platelet aggregate formation, the acquired sequential images were first denoised with NIS-Elements Advanced Research (AR) version 5.21.00 (Nikon). Images with vascular damage areas of 25–40 μm for venous bleeding and 10–25 μm for arterial bleeding were then extracted. Platelet, fibrin, and plasma signals were quantified via NIS-Elements AR. The height of adhered platelet aggregates from the endothelial end to the luminal end, defined as the platelet height, was measured (Figs. 7 and 8). The protrusion of platelet aggregates outside the vessel was assessed by their length beyond the vascular endothelium (Fig. 8). Fibrin was quantified by the area of fibrin formed (Figs. 7 and 8), which was determined by the stronger signals compared with circulated blood, and bleeding was measured by the leaked blood cells and the signal of leaked plasma. The data were analysed via Microsoft® Excel for Mac version 16.79.1. The maximum platelet height was defined as the peak height.
For bleeding analysis, the maximum intensity of the leaked plasma signal was normalized to 1. In veins, the area under the curve (AUC) of the leaked plasma signal was calculated from 1 to 181 s; in arteries, it was calculated from the time of a 0.4 bleeding signal to 246 s. The fibrin signals in the veins were normalized to a minimum of 0 and a convergence value of 1. For FVIII KO mice, a quadratic fitting curve was generated from values above 0. Other strains’ signals from 0 to 120 s were used to create quadratic fitting curves. The slope of the tangent line for a signal of 0.5 was calculated for these curves. In arteries, a quadratic fitting curve was created from fibrin signals ranging from 0.2 to 0.8, and the slope for 0.5 was similarly calculated. The time for blood cells to stop leaking was confirmed via video.
Statistics and Reproducibility
Statistical analyses were conducted via RStudio version 2023.12.0 + 369, with R version 4.3.2 (R Foundation for Statistical Computing, Vienna, Austria). The Wilcoxon signed-rank test was used to compare matched pairs via the ‘exactRankTests’ package (version 0.8-35), whereas the Steel–Dwass test with the Monte Carlo method for three or more independent groups was performed via the ‘NSM3’ package (version 1.18). P values < 0.05 indicated statistical significance.
Responses