Generation of live mice from haploid ESCs with germline-DMR deletions or switch
Introduction
In the 1980s, the bi-maternal and bi-paternal embryos were constructed by pronuclear transfer. These embryos exhibited developmental failure during mid-gestation, leading to the discovery of genomic imprinting1,2,3, as parental genomes bear distinct “marks” on their DNA. Genomic imprinting is an epigenetic process that causes a subset of genes to be monoallelically expressed in a parent-of-origin-specific manner. Most imprinted genes are organized in clusters, and their monoallelic expression is regulated by the parent-of-origin-specific acquisition of DNA methylation marks, termed the differentially methylated regions (DMRs)4,5,6,7. Genetic studies have demonstrated that DMR deletions could largely mimic their hypermethylated state in most loci, resulting in expected imprinting patterns during embryonic development8,9,10. Extensive research has highlighted the critical roles of imprinted genes during embryonic and fetal development6,7,8. However, the specific sets of paternal and maternal imprinted genes essential for development remain unclear, largely due to the lack of an efficient system to genetically manipulate imprinted control regions of both paternally and maternally imprinted loci.
Recently, mammalian haploid embryonic stem cells (haESCs) have been successfully created from blastocysts carrying only one set of maternal or paternal genome11,12,13,14,15,16. Notably, androgenetic-haESCs (AG-haESCs), derived from sperm-originated haploid blastocysts, could support full-term embryonic development when injected into oocytes, resulting in healthy animals, termed semi-cloned mice (SC mice)12. However, AG-haESCs gradually lost the potential to produce SC animals because two inhibitors of Mek1/2 and Gsk3β (2i) used in their culture medium induced a widespread loss of DNA methylation, including H19-DMR and IG-DMR12,17. Strikingly, AG-haESCs carrying both H19-DMR and IG-DMR deletions could efficiently support the development of SC embryos18, demonstrating that DMR deletions can simulate the paternal imprinting state. Similarly, parthenogenetic-haESCs (PG-haESCs), derived from haploid embryos carrying oocyte genomes and cultured in 2i medium, also lost DMR methylation over time, and this loss enabled these cells to produce SC mice after removal of both H19-DMR and IG-DMR19,20. Together, imprint-free haESCs with paternal DMR deletions can be used as sperm replacement to support embryonic development, implying that these cells with particular maternal DMR deletions may also be used as oocyte genome replacement to support embryonic development. We thus reasoned that replacing both sperm and oocyte genomes with haESC-derived DNA could offer a novel strategy to manipulate paternal and maternal imprints simultaneously, facilitating the identification of essential imprinting combinations for embryonic development. Moreover, this strategy might also enable systematic studies on interactions between paternal and maternal imprints.
In this study, we tested this concept by establishing a fusion system between AG-haESC and PG-haESC lines with parent-of-origin-specific-DMR edits (Ha-Ha-fusion system) to produce diploid ESCs carrying DMR-deletion combinations. These cells were then subjected to tetraploid (4N) embryo complementation to assess their developmental potential. The 4N embryo complementation method, widely utilized to rescue the placental dysfunction that otherwise would result in fetal death21,22, allows 4N cells to contribute extraembryonic tissues while rarely contributing to the embryo itself23,24. This strategy has been used to assess the pluripotency of ESCs and iPSCs, leading to the generation of all-ESC or all-iPSC mice25,26. Here, we demonstrated that a deletion combination of 10 regions, including paternal H19-DMR and IG-DMR, and maternal Xist-DMR, Igf2r-DMR, Kcnq1-DMR, Gnas-DMR, Peg3-DMR, Nespas-DMR, Snrpn-DMR, and Grb10-DMR could support reconstructed embryos to accomplish full-term embryonic development efficiently. Furthermore, we showed that the non-parent-of-origin-specific inheritance mode of DMR deletions could also support embryonic development, indicating that their primary functional role lies in regulating the dosage of imprinted genes rather than their parental origin.
Results
Establishment of Ha-Ha-fusion system based on hypomethylated haESCs induced by 2i
To comprehensively investigate the functional significance of DMRs in vivo, we aimed to establish a system capable of simultaneously manipulating maternal and paternal DMRs. To achieve this, we first derived RFP-labeled AG-haESCs (RFP-AG-haESCs) and GFP-labeled PG-haESCs (GFP-PG-haESCs) from CAG-RFP and actin-EGFP transgenic mice respectively27,28 (Supplementary Fig. S1a). The haESCs were then cultured in a standard ESC culture medium supplemented with 2i and maintained through FACS-enrichment of haploid cells regularly12 (Fig. 1a, b). Given that 2i induces global hypomethylation following passaging, as reported previously17,29,30, whole-genome bisulfite sequencing (WGBS) on GFP-PG-haESCs and RFP-AG-haESCs at passage 30 (P30) was performed. This analysis revealed that both haploid cell lines exhibited global hypomethylation compared to oocyte and sperm31 (Fig. 1c; Supplementary Fig. S1b–g). Notably, all paternal DMRs and the vast majority of maternal DMRs were hypomethylated (Fig. 1d). Subsequently, RFP-AG-haESCs and GFP-PG-haESCs were fused using HVJ virus, and the diploid (2N) fused cells with both GFP and RFP signals were enriched by FACS (termed Ha-Ha-fusion system, Fig. 1e). To assess their imprinted state, we analyzed the methylation state of 2N ESCs fused using PG-haESCs at P50 and found that the fused ESCs exhibited DNA methylation levels between AG-haESCs and PG-haESCs (Fig. 1c; Supplementary Fig. S1b–g) and sustained hypomethylated state at imprinted regions (Fig. 1d). We further investigated the methylation status of DMRs in haploid ESCs and 2N fused cells across different passages using bisulfite PCR. Consistently, RFP-AG-haESCs (P30) displayed hypomethylation at H19-DMR and IG-DMR (Fig. 1f), and GFP-PG-haESCs (P30) retained methylation at a reduced level at Snrpn-DMR and Peg3-DMR (Supplementary Fig. S1h). However, by P50, GFP-PG-haESCs lost all DNA methylation at Snrpn-DMR and Peg3-DMR (Fig. 1f). Similarly, 2N fused ESCs showed a near-complete loss of DNA methylation at two tested DMRs by P50 (Fig. 1g). Together, these findings indicate that haESCs and 2N fused ESCs exhibit imprinted-free state after long-term culture under a condition with 2i.
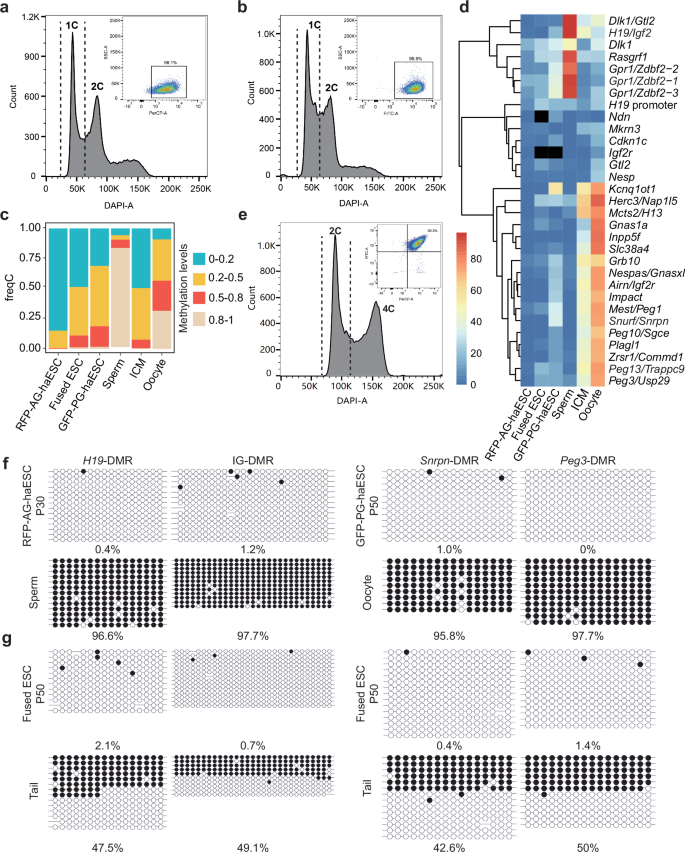
a Establishment of RFP-labeled AG-haESCs by FACS enrichment for haploid cells. A DAPI filter was used to detect a signal of Hoechst 33342-stained DNA. A PerCP-A filter was used to detect a signal of RFP-positive cells (98.1%). b Establishment of GFP-labeled PG-haESCs by FACS enrichment for haploid cells. A DAPI filter was used to detect a signal of Hoechst 33342-stained DNA. A FITC-A filter was used to detect a signal of GFP-positive cells (99.8%). c Methylation levels of the whole genome in oocytes, sperm, inner cell mass (ICM), RFP-labeled AG-haESCs, GFP-labeled PG-haESCs and diploid fused ESCs. DNA methylome data of oocytes, sperm and ICM have been reported31. d Heatmap of imprinted region methylation in oocytes, sperm, ICM, RFP-labeled AG-haESCs, GFP-labeled PG-haESCs and diploid fused ESCs. Black squares indicate a lack of valid reads. e Establishment of diploid fused ESCs. A DAPI filter was used to detect a signal of Hoechst 33342-stained DNA. FITC-A filter and PerCP-A filter were used to detect a signal of double-positive cells (99.3%). f Bisulfite sequencing analysis of IG-DMR and H19-DMR in P30 RFP-labeled AG-haESCs and sperm, and Snrpn-DMR and Peg3-DMR in P50 GFP-labeled PG-haESCs and oocyte. Open and filled circles represent unmethylated and methylated CpG sites, respectively. g Bisulfite sequencing analysis of IG-DMR, H19-DMR, Snrpn-DMR and Peg3-DMR in P50 fused ESCs and tail. Open and filled circles represent unmethylated and methylated CpG sites, respectively. The passage number of fused ESCs is inherited from the passage number of PG-haESCs.
Embryos reconstructed from Ha-Ha-fusion-derived diploid ESCs with paternal DKO and six maternal DMR deletions develop to E15.5
We next investigated whether all-haESC mice can be generated through injection of 2N ESCs fused between RFP-AG-haESCs and GFP-PG-haESCs with DMR deletions into tetraploid embryos (Fig. 2a). To this end, H19-DMR and IG-DMR were deleted in RFP-AG-haESCs (termed RFP-DKO) to mimic paternal inherited imprints (Supplementary Fig. S2a). To evaluate the feasibility of the Ha-Ha-fusion system, wild-type GFP-PG-haESCs (GFP-WT) at different passages were fused with RFP-DKO cells. The resulting 2N fused ESCs (WT-DKO) carrying both GFP and RFP markers were enriched by FACS and their developmental potential was assessed through tetraploid complementation (Fig. 2b). After transplantation of reconstructed embryos into uteruses of recipient females, WT-DKO cells (P8) derived from early-passage GFP-WT cells (P5) could support full-term embryonic development with a birth rate at around 15%, half of which survived to adulthood (Table 1; Supplementary Fig. S2b, c). By contrast, the reconstructed embryos derived from WT-DKO cells (P30) fused between RFP-DKO and late-passage GFP-WT cells (P27) were completely arrested at E10.5 (Fig. 2c). These results highlight the importance of maternal imprints, as their absence results in developmental failure of reconstructed embryos before E10.5.
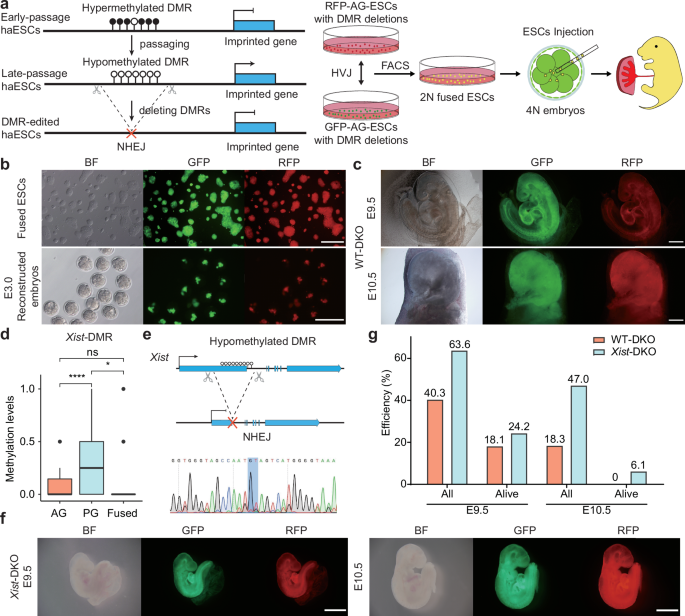
a Scheme of generating embryos with parental-allele-specific DMR deletions. Hypomethylated DMRs were deleted by CRISPR-Cas9 in RFP-labeled AG-haESCs and GFP-labeled PG-haESCs. Diploid fused cells were obtained by fusing RFP-labeled AG-haESCs with GFP-labeled PG-haESCs with HVJ virus. 2N double-positive cells were enriched by FACS. Reconstructed embryos were generated by tetraploid complementation of fused cells. b Colony morphology and fluorescence labeling of diploid fused ESCs (top) and E3.0 reconstructed embryos (bottom). Scale bars, 200 µm. BF, bright field. c E9.5 (top) and E10.5 (bottom) reconstructed embryos (WT-DKO) carrying paternal H19-DMR and IG-DMR deletions. WT-DKO embryos were generated from late passages (P30) fused ESCs. The passage number of fused ESCs is inherited from the passage number of PG-haESCs. Scale bars, 500 µm. d DNA methylation levels at Xist-DMR in AG-haESCs, PG-haESCs and fused ESCs. Data are presented as mean ± SD, analyzed by Student’s t-test, *P < 0.05, ****P < 0.0001, ns, not significant. e Top, diagram showing deletion of Xist-DMR. Bottom, DNA sequences of PCR products amplified from Xist-DMR’s deleted region in Xist-DMR deletion PG-haESCs. f E9.5 (left) and E10.5 (right) reconstructed embryos (Xist-DKO) carrying paternal DKO and maternal Xist-DMR deletion. Scale bars, 1 mm. g A comparison of the developmental (the number of embryos in the amniotic sac divided by the number of embryos transferred) and alive rates of reconstructed embryos in c and f. Alive status is judged by the heartbeat. Data were generated from Table 1.
To identify maternal DMRs critical for embryonic development, we sought to enhance the developmental potential of reconstructed embryos by deleting maternal DMRs in GFP-PG-haESCs. Previous studies on somatic cell nuclear transfer (SCNT)-derived cloned embryos indicated that ectopic expression of maternal Xist contributed to implantation defects32. Reducing Xist dosage, either by using Xist heterozygous knockout donor cells or injecting small interfering RNA (siRNA) against Xist into one-cell cloned embryos, significantly increased cloning efficiency by 8- to 9-fold32,33. Given that haESCs carry only X chromosome12 and 2N fused ESCs are female cells with XX (Supplementary Fig. S2d), we thus reanalyzed WGBS results and found that 2N fused ESCs completely lost all DNA methylation at the Xist promotor (DMR) (Fig. 2d), implying that aberrant X-chromosome inactivation (XCI) may be involved in developmental failure of reconstructed embryos from 2N WT-DKO ESCs. To address this, we deleted Xist-DMR in GFP-WT cells (termed GFP-Xist-KO) (Fig. 2e), and then fused them with RFP-DKO cells to produce a stable diploid cell line (termed Xist-DKO). Tetraploid complementation experiments revealed that Xist-DMR deletion in GFP-WT dramatically improved post-implantation developmental rate of Xist-DKO embryos at E10.5 (47.0% compared to 18.3% of WT-DKO embryos, Table 1). Moreover, 6.1% of transferred embryos remained viable at E10.5 (Fig. 2f, g). These results indicate that correction of Xist dosage can dramatically rescue embryonic development of the reconstructed embryos.
To further improve the developmental potential of reconstructed embryos, we targeted additional maternal loci essential during mid-gestation by deleting Igf2r- and Kcnq1-DMRs in GFP-Xist-KO cells, whose deletion caused embryonic lethality before E1534,35,36,37. The resultant cells (termed GFP-XIK) were fused with RFP-DKO cells to produce diploid cells carrying five DMR deletions (termed XIK-DKO) (Supplementary Fig. S2e), which could develop to E13.5 through tetraploid complementation (Table 1; Supplementary Fig. S2f, g), but all arrested at E14.5 (Table 1; Supplementary Fig. S2g).
To restore maternally imprinted states critical for late gestation, we deleted additional DMRs in GFP-XIK cells. Previous studies showed that imprinting abnormalities in Nespas/Gnas locus led to developmental deficiency at late gestation38,39,40,41,42, while Peg3 locus modulates fetal growth at late gestation43 and its aberrant expression causes partial embryonic lethality and reduced body weight44. Thus, Gnas-DMR and Peg3-DMR were deleted in the cell line GFP-XIK (termed GFP-XIKGP) (Supplementary Fig. S2e). In vivo developmental potency analysis indicated that GFP-XIKGP cells, when fused with RFP-DKO cells (XIKGP-DKO), extended embryonic development to E14.5 (2.5%) (Table 1; Supplementary Fig. S2h, i). Furthermore, after removal of Nespas-DMR in GFP-XIKGP cells to produce a cell line termed GFP-XIKGPN (Supplementary Fig. S2e), we found that the reconstructed embryos (XIKGPN-DKO) could further develop to E15.5 (Supplementary Fig. S2j), though no embryos (0/90) survived to E17.5 (Table 1; Supplementary Fig. S2j, k). In conclusion, the deletion of 6 maternal DMRs critical for embryonic development in PG-haESCs improved the developmental potential of reconstructed embryos, extending their viability from E9.5 to E15.5.
Efficient generation of live pups using Ha-Ha-fusion-derived diploid ESCs with two more maternal DMR deletions
A recent study showed that Snrpn-DMR and Grb10-DMR deletions contribute to the full-term development of bi-paternal embryos45. Snrpn locus is associated with prenatal growth, and its abnormalities lead to multiple postnatal phenotypes, including neonatal hypotonia and failure to thrive46,47,48. To investigate its role, we first deleted Snrpn-DMR in the GFP-XIKGPN cells to produce a cell line with 7 maternal DMR deletions (termed GFP-XIKGPNS) (Supplementary Fig. S2e, l). Following fusion with RFP-DKO cells, 4N complementation analysis showed that 5.1% of the resultant reconstructed embryos (termed 7KO-DKO) were viable at E19.5 (Fig. 3a and Table 1; Supplementary Fig. S2m). However, all pups died shortly after birth. These findings indicate that the re-establishment of maternal Snrpn-DMR enables full-term development of reconstructed embryos, suggesting a critical role of Snrpn-imprinted region during perinatal stages.
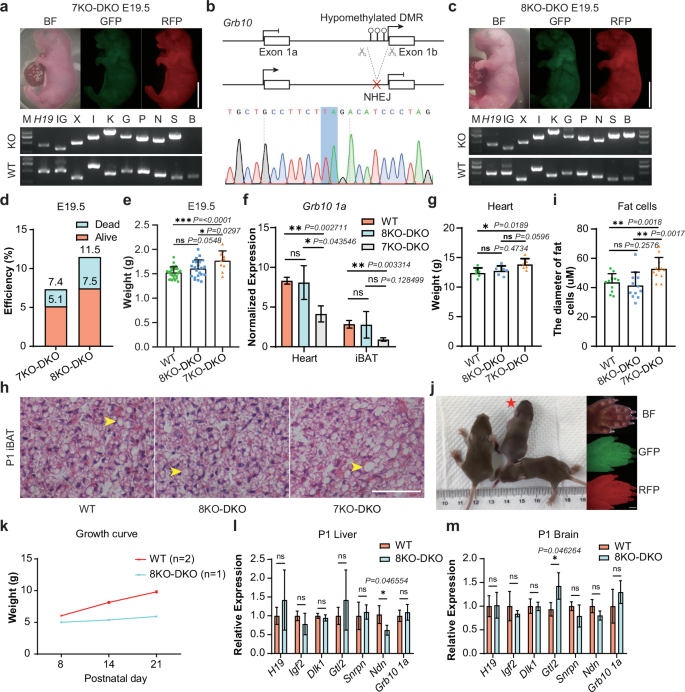
a Top, E19.5 reconstructed embryos (7KO-DKO) carrying paternal DKO and maternal 7KO. 7 maternal imprinted regions are Xist (X), Igf2r (I), Kcnq1 (K), Gnas (G), Peg3 (P), Nespas (N) and Snrpn (S). Bottom, Genotyping of the 7KO-DKO ESCs with PCR. Genotypes of the deletion and WT were checked for paternal DKO and maternal 7KO. Scale bar, 5 mm. b Top, diagram showing deletion of Grb10-DMR. Bottom, DNA sequences of PCR products amplified from Grb10-DMR’s deleted region in GPF-XIKGPNSB PG-haESCs. c Top, E19.5 reconstructed embryos (8KO-DKO) carrying paternal DKO and maternal 8KO. 8 maternal regions are 7 maternal regions in a and Grb10-DMR (B). Bottom, genotyping of the 8KO-DKO ESCs with PCR. Genotypes of the deletion and WT were checked for paternal DKO and maternal 8KO. Scale bar, 5 mm. d Development rate of 7KO-DKO and 8KO-DKO embryos at E19.5. Alive status is judged by the breath. Data were generated from Table 1. e The body weights of E19.5 BDF1 (WT, n = 30), 8KO-DKO (n = 22) and 7KO-DKO (n = 10) embryos. Data are presented as mean ± SD, analyzed by Student’s t-test, *P < 0.05, ***P < 0.001, ns, not significant. f Expression analysis of Grb10 1a in hearts and iBATs of P1 WT, 8KO-DKO and 7KO-DKO pups (n = 3 for each group). Data are presented as mean ± SD, analyzed by Student’s t-test, *P < 0.05, **P < 0.01, ns, not significant. g The weights of heart from P1 WT (n = 6), 8KO-DKO (n = 6) and 7KO-DKO (n = 6) pups. Data are presented as mean ± SD, analyzed by Student’s t-test, *P < 0.05, ns, not significant. h H&E staining of iBATs from P1 WT, 7KO-DKO and 8KO-DKO mice. Scale bar, 400 µm. Multilocular lipid droplets were marked by yellow arrows. i Average diameters of fat cells (n = 12) from iBATs in P1 WT, 8KO-DKO and 7KO-DKO pups. Data are presented as mean ± SD, analyzed by Student’s t-test, **P < 0.01, ns, not significant. j Left, postnatal 23 day mice of WT and 8KO-DKO. 8KO-DKO pup was marked by a red asterisk. Right, fluorescent images of 8KO-DKO pup’s forepaw. Scale bar, 5 mm. k The growth curve of WT (n = 2) and 8KO-DKO (n = 1) mice. l, m Expression analysis of imprinted genes (H19, Igf2, Dlk1, Gtl2, Snrpn, Ndn and Grb10 1a) in liver (l) and brain (m) of P1 WT and 8KO-DKO pups (n = 3 for each group). Data are presented as mean ± SD, analyzed by Student’s t-test, *P < 0.05, ns, not significant.
Next, we targeted the Grb10 locus, which is associated with lipid biogenesis and regulation of social behaviors49,50. We deleted Grb10-DMR in the GFP-XIKGPNS cells to produce GPF-XIKGPNSB cells (Fig. 3b; Supplementary Fig. S2n). Remarkably, 11.5% (37/322) of the resultant reconstructed embryos (termed 8KO-DKO) survived to E19.5 (Fig. 3c, d), with 64.9% (24/37) of the embryos remaining viable (Fig. 3d and Table 1). Interestingly, the body weights of 8KO-DKO-originated pups were lighter than those of 7KO-DKO pups but similar to WT (BDF1) female pups (Fig. 3e). These results indicate that the deletion of Grb10-DMR mitigates the overgrowth of 7KO-DKO embryos, coincident with a previous study showing that Grb10 1a expression inhibited growth51. Accordingly, the Grb10 1a expression was up-regulated in tested tissues of 8KO-DKO mice compared to that in 7KO-DKO embryos (Fig. 3f), suggesting that Grb10-DMR deletion effectively simulates the methylated allele. Further analysis revealed that the hearts of 7KO-DKO mice were heavier than those of the WT mice, whereas no significant differences in heart weight were observed between 8KO-DKO and WT pups (Fig. 3g; Supplementary Fig. S3a, b). Although the weights of interscapular brown adipose tissue (iBAT) were comparable across groups, histological analysis using hematoxylin and eosin (H&E) staining showed larger multilocular lipid droplets in iBAT of the 7KO-DKO pups at postnatal day 1 compared to the WT and 8KO-DKO mice (Fig. 3h, i; Supplementary Fig. S3c). These results are consistent with previous observations that Grb10 1a is critical for the development of the heart and iBAT50,52.
While normal-weight pups were successfully fostered, most died within a few days of birth (Fig. 3j; Supplementary Fig. S3d, e). Two pups survived over one week; one of the pups that survived displayed obvious growth retardation and died at postnatal day 8 (Supplementary Fig. S3e, f), while the other failed to thrive and died at postnatal day 23 (Fig. 3j, k). Gene expression analysis revealed that although DMR deletions partially rescued the expression of genes located in these imprinted regions, several critical genes remained aberrantly expressed during development (Fig. 3l, m; Supplementary Fig. S3g, h). Furthermore, WGBS analysis of tail samples from postnatal day 23 8KO-DKO mice (D23) and WT mice (BDF1) showed that the majority of DMRs, including all deleted regions, were hypomethylated in the D23 sample (Supplementary Fig. S4a, b). However, secondary DMRs sustain a high methylation level in the D23 sample, similar to that in the WT sample (Supplementary Fig. S4c, d), suggesting that the deletion of germline DMRs can induce the establishment of the DNA methylation status in secondary DMRs during development. Taken together, while DMR deletions do not fully restore normal expression patterns of imprinted gene clusters, parental-allele-specific deletion of 10 DMRs can efficiently support the full-term development of reconstructed embryos.
Embryos reconstructed from Ha-Ha-fusion-derived diploid ESCs with paternally-derived Snrpn-DMR deletion can develop to term
Having demonstrated that DMR-deletion combinations in haploid cells can mimic parental imprinting state and support full-term embryonic development, we next investigated whether parental origins of germline DMRs would affect their function on development by changing the location of Snrpn-DMR deletion. To this end, we deleted the Snrpn-DMR in the RFP-DKO cells to mimic paternally-derived Snrpn imprinting (termed RFP-DKO-S) and fused these cells with GFP-XIKGPNB cells (Fig. 4a). Remarkably, 4N complementation analysis showed that the resultant cells (termed 7KO-DKO + S) supported full-term embryonic development achieving a developmental state comparable to that of fused cells with maternally derived Snrpn-DMR deletion (8KO-DKO) (Fig. 4b, c). By contrast, XIKGPNB-DKO embryos generated from fused cells between RFP-DKO and GFP-XIKGPNB exhibited growth-retarded phenotype at E17.5, highlighting the critical role of paternal Snrpn-DMR deletion in enabling full-term development of reconstructed embryos (Fig. 4d, e). Additionally, embryos derived from 7KO-DKO + S cells had similar body weights to 8KO-DKO embryos (Fig. 4f). These results indicated that paternal transmission of Snrpn-DMR deletion extends the developmental potential of fused cells lacking Snrpn-DMR deletion from E17.5 to birth.
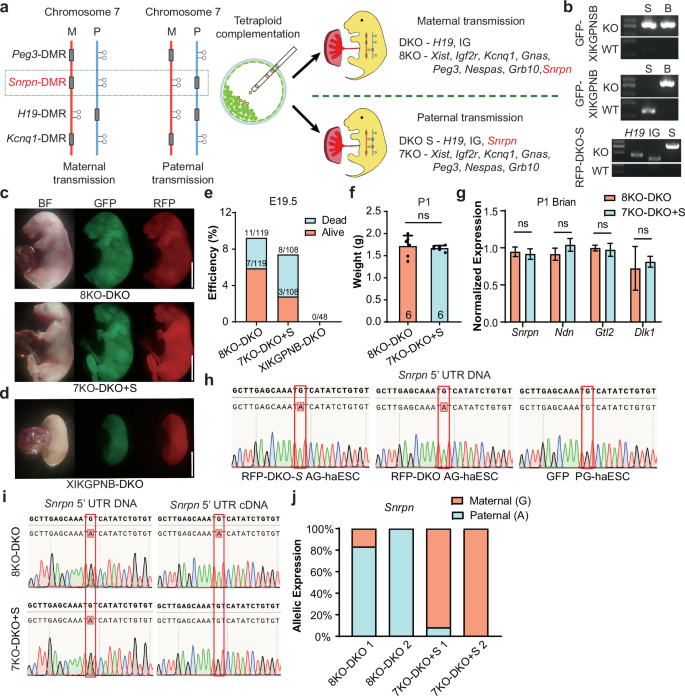
a Schematic diagram showing the generation of reconstructed embryos (7KO-DKO + S) carrying paternally derived Snrpn-DMR deletion. Snrpn-DMR was deleted in RFP-DKO AG-haESCs (termed RFP-DKO-S), and Xist-DMR, Igf2r-DMR, Kcnq1-DMR, Gnas-DMR, Peg3-DMR, Nespas-DMR and Grb10-DMR were deleted in PG-haESCs (termed GFP-XIKGPNB). b Genotyping of the related cell lines with PCR. Genotypes of GFP-XIKGPNSB PG-haESCs, GFP-XIKGPNB PG-haESCs and RFP-DKO-S AG-haESCs were indicated on the top, middle and bottom gel images, respectively. c Left, fluorescent images of E19.5 8KO-DKO pups. Right, fluorescent images of E19.5 7KO-DKO + S pups. Scale bars, 5 mm. d Fluorescent images of E17.5 XIKGPNB-DKO embryos. Scale bar, 5 mm. e Development rate of 8KO-DKO, 7KO-DKO + S and XIKGPNB-DKO embryos at E19.5. Alive status is judged by the breath. f The body weights of P1 8KO-DKO (n = 6) and 7KO-DKO + S pups (n = 6). Data are presented as mean ± SD, analyzed by Student’s t-test, ns, not significant. g Expression analysis of Snrpn, Ndn, Gtl2 and Dlk1 in brain of E19.5 8KO-DKO and 7KO-DKO + S embryos (n = 3 for each group). Data are presented as mean ± SD, analyzed by Student’s t-test, ns, not significant. h Snrpn 5’ UTR DNA sequences of PCR products amplified from DNA of RFP-DKO-S AG-haESCs (top), RFP-DKO AG-haESCs (middle) and PG-haESCs (bottom). i Left, Snrpn 5’ UTR DNA sequences of PCR products amplified from tail DNA of 8KO-DKO (top) and 7KO-DKO + S (bottom) embryos. Right, Snrpn 5’ UTR DNA sequences of PCR products amplified from brain cDNA of 8KO-DKO (top) and 7KO-DKO + S (bottom) embryos. j The relative allelic expression of the Snrpn, as measured by Snrpn 5’ UTR SNP analysis from brains cDNA of 8KO-DKO and 7KO-DKO + S embryos.
To further validate these results, we analyzed the expression patterns of the typical genes in the brain, where both IG-DMR and Snrpn-DMR play vital roles during the perinatal development stage. The expression levels of all tested genes in both imprinted clusters were comparable between 7KO-DKO + S mice and 8KO-DKO mice (Fig. 4g). Since the single nucleotide polymorphisms (SNPs) lack in Snrpn transcripts between RFP-AG-haESCs and GFP-PG-haESCs, to analyze the parental origins of gene expression, we introduced an adenine to replace guanine in 5’ LTR of Snrpn in RFP-AG-haESCs to distinguish its expression from maternal expression of GPF-PG-haESCs carrying the guanine at the same site (Fig. 4h). As expected, Snrpn was expressed from the paternal allele in the brain of 8KO-DKO mice as a maternally imprinted gene and expressed from maternal allele in 7KO-DKO + S mice as a paternally imprinted gene (Fig. 4i, j). Together, these results show that the maternally or paternally-derived Snrpn-DMR deletion functions equivalently during embryonic development of reconstructed embryos, suggesting that dosage regulation, rather than parental origin, is the key of imprinted genes.
SC embryos with maternally-derived H19-DMR deletion can develop into adults with an inverted expression of the imprinted gene
We next investigated whether paternally methylated H19-DMR could also function properly through maternal transmission. Our previous studies have shown that the majority of SC embryos generated by injection of WT AG-haESCs into oocytes exhibited growth-retarded phenotypes due to loss of H19-DMR and IG-DMR methylation12. Furthermore, it was shown that deletion of both H19-DMR and IG-DMR in AG-haESCs (DKO-AG-haESCs) enabled efficient support of full-term SC embryo development18. Based on this, we tested whether SC embryos generated by injection of AG-haESCs with IG-DMR deletion into oocytes with H19-DMR deletion could develop into viable animals. To achieve this, we first generated a mouse line carrying homozygous H19-DMR deletions (termed H19-KO mice) through breeding SC mice generated from DKO-AG-haESCs. Subsequently, we deleted IG-DMR in the hypomethylated RFP-AG-haESCs (termed RFP-IG-KO AG-haESCs) (Fig. 1f). Next, RFP-IG-KO AG-haESCs were injected into mature oocytes with H19-DMR deletion to produce SC embryos with paternally-derived IG-DMR deletion and maternally-derived H19-DMR deletion (termed H19-IG embryos) (Fig. 5a; Supplementary Fig. S5a). SC embryos were collected at E12.5 to assess development, as a previous study highlighted the critical role of H19-DMR during early embryogenesis53. The results showed that H19-IG embryos exhibited developmental features comparable to those of SC embryos obtained by injection of RFP-DKO-AG-haESCs into WT oocytes (WT-DKO embryos) (Table 2; Supplementary Fig. S5b–d). By contrast, SC embryos derived from the injection of AG-haESCs with only IG-DMR deletion into WT oocytes (WT-IG embryos) were degenerative at E12.553. These results indicated that maternal deletion of H19-DMR plays critical roles for the development of H19-IG embryos at E12.5. Consistent with this, the expression levels of genes located in this imprinted locus were overall similar between the fetus and placenta of E12.5 H19-IG embryos and E12.5 WT-DKO embryos (Supplementary Fig. S5e–h). However, H19 expression was lower in the fetus of H19-IG embryos (Supplementary Fig. S5e), potentially due to residual H19-DMR methylation in some SC embryos30. Moreover, we dissected H19-IG embryos at E16.5 and found that they also displayed developmental characteristics as WT-DKO embryos (Table 2; Supplementary Fig. S5i–n).
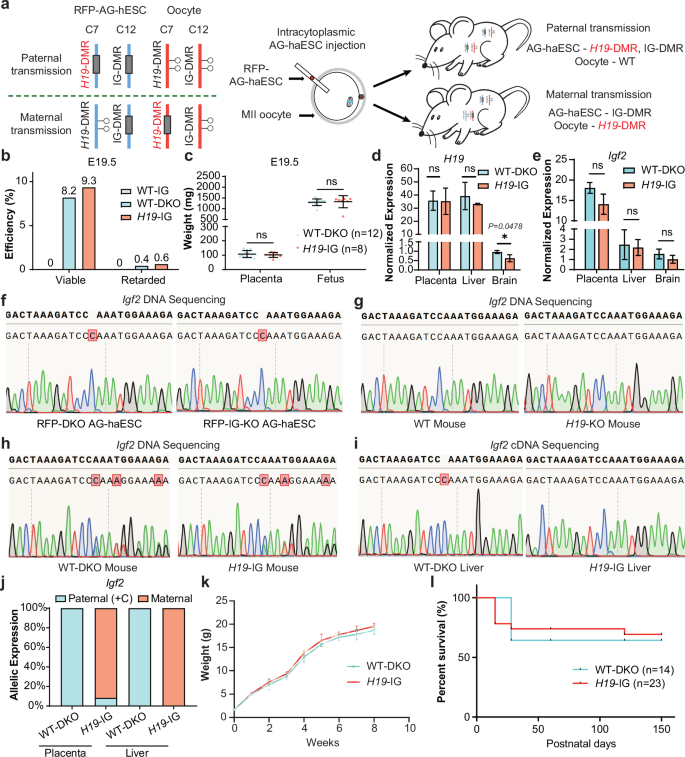
a Schematic diagram showing generation of SC embryos (H19-IG embryos) carrying maternally-derived H19-DMR deletion. H19-DMR was deleted in oocytes, which derived from H19-KO mice. IG-DMR was deleted in RFP AG-haESCs (RFP-IG-KO). Reconstructed embryos were obtained by intracytoplasmic AG-haESCs injection (ICAHCI). b A comparison of the development rates of E19.5 WT-IG, WT-DKO and H19-IG embryos. Viable status is judged by the breath. Data were generated from Table 2. c Fetal and placental weights of E19.5 WT-DKO (n = 12) and H19-IG (n = 8) embryos. Data are presented as mean ± SD, analyzed by Student’s t-test, ns, not significant. d, e Expression analysis of H19 (d) and Igf2 (e) in placenta, liver and brain of P1 WT-DKO and H19-IG pups (n = 3 for each group). Data are presented as mean ± SD, analyzed by Student’s t-test, *P < 0.05, ns, not significant. f–i Igf2 5’ UTR DNA sequences of PCR products amplified from DNA of RFP-DKO and RFP-IG-KO AG-haESCs (f), tail DNA of WT and H19-KO mouse (g), tail DNA of WT-DKO and H19-IG mouse (h), liver cDNA of WT-DKO and H19-IG mouse (i). j Relative allelic expression of the Igf2, as measured by Igf2 5’ UTR SNP analysis from placenta and liver cDNA of WT-DKO and H19-IG pups. k Growth curve of WT-DKO (n = 5) and H19-IG (n = 5) mice. l Survival curve of WT-DKO (n = 14) and H19-IG (n = 23) mice.
Notably, 46 alive H19-IG mice were produced from 492 transferred embryos, achieving a developmental efficiency of 9.3%, similar to that of WT-DKO mice (8.2%, 19/232, Fig. 5b and Table 2; Supplementary Fig. S5o). Consistently, they had similar weights and gene expressions as WT-DKO pups (Fig. 5c–e). To confirm the parental origin of transcripts, we introduced one cytosine in 5′-LTR of Igf2 into RFP AG-haESCs (Fig. 5f, g). As expected, the resultant H19-IG embryos expressed maternally-biased expressed Igf2 in all tested tissues (Fig. 5h–j). Moreover, H19-IG mice showed normal postnatal growth (Fig. 5k, l). These findings demonstrate that maternal transmission of H19-DMR deletion could support the normal development of SC embryos. Collectively, our findings indicate that non-parent-of-origin-specific inheritance mode of DMRs is capable of supporting embryonic development normally, implying that the primary function of DMRs is to regulate the expression dosage of imprinted genes rather than their parental origin.
Discussion
Parental-origin-specific genomic imprinting is critical for mammalian development6,7. However, systematic studies on the functional significance of imprinting networks have been limited due to the absence of an efficient strategy to simultaneously manipulate both maternal and paternal imprints. In this study, we showed that the combined application of Ha-Ha-fusion and 4N embryo complementation methods allows a stepwise restoration of parental imprints for embryonic development, enabling the generation of live all-haESC-derived pups. These results highlight that 10 imprinted clusters, including two paternally imprinted loci, H19–Igf2 and Dio3–Dlk1, and eight maternally imprinted loci, Xist, Airn–Igf2r, Kcnq1, Gnas, Peg3, Nespas–Gnasxl, Snrpn, and Grb10, are sufficient for full-term embryonic development. Our results confirm that monoallelic expression of imprinted genes is essential for embryonic and fetal development. Additionally, we showed that germline DMR deletions can be switched between the paternal and maternal genome, emphasizing that it is the monoallelic expression of imprinted genes, rather than their parental origin, that is crucial for proper development and growth.
Here, we fixed the paternal imprinted state using AG-haESCs with both H19-DMR and IG-DMR deletions and investigated the appropriate maternal imprinted state for embryonic development using PG-haESCs with different maternal DMR deletions. Through a stepwise strategy, we systematically analyzed the relationship between maternal DMRs and embryonic development. Our results indicate that maternal deletion of Xist-DMR can improve the developmental rate of reconstructed embryos without maternal DMR deletions (WT-DKO) from E9.5 to E10.5; Igf2r-DMR and Kcnq1-DMR deletions can improve the developmental potential to E13.5; Gnas-DMR and Peg3-DMR deletions can improve the developmental potential to E14.5; Nespas-DMR deletion can improve the developmental potential to E15.5; Snrpn-DMR deletion can improve the developmental potential to E19.5; and strikingly, Grb10-DMR deletion allows pups to survive up to postnatal day 23 (Fig. 6a).
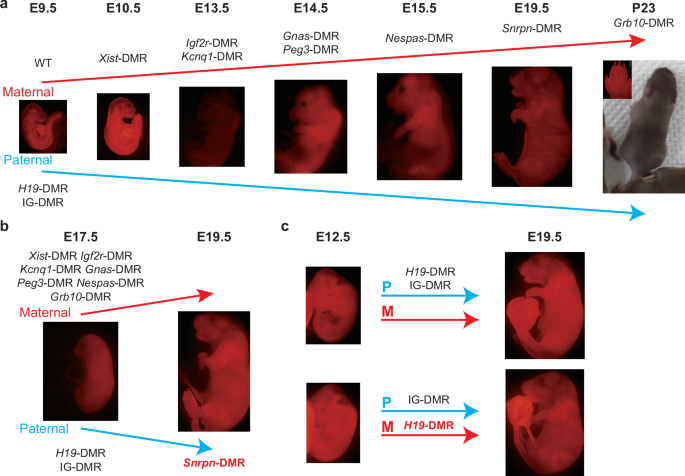
a Schematic diagram showing generation of reconstructed embryos carrying 8 maternal DMR deletions and 2 paternal DMR deletions. Developmental potential of reconstructed embryos was improved from E9.5 to postnatal day 23. b, c Schematic diagram showing that Snrpn-DMR (b) and H19-DMR (c) with inverse parental origins can regulate proper embryonic growth.
Interestingly, a recent study reported that AG-haESCs with the same maternal DMR deletions except Xist-DMR could be used to replace the oocyte genome to support bi-paternal development45. However, the birth rate of bi-paternal embryos was quite low (around 1% of transferred embryos) and all pups died a few days after birth. By contrast, incorporating Xist-DMR deletion in our strategy significantly increased birth rate of reconstructed embryos and improved postnatal development. This aligns with previous studies that Xist dosage balance is critical for embryonic development at mid-gestation54 and the failure of random XCI in the embryo proper also caused mice to be born at a lower frequency and smaller in size55. Additionally, SCNT studies have shown that the correction of skewed XCI pattern significantly improved the developmental efficiency of cloned embryos32,33. Taken together, these results indicate Xist dosage balance is critical for mid-gestation development, which is a foundation for later developmental stages.
Our data further demonstrated that DMRs with inverse parental origins can regulate embryonic development normally (Fig. 6b, c). This is consistent with reports showing that parental-origin inversion of gene expression in the Dlk1–Dio3 locus can support normal development through generation of a mouse model carrying paternal deletion of IG-CGI and maternal deletion of IG-DMR10. Similarly, maternal expression of Rasgrf1 or Igf2, normally expressed from the paternal allele, rescues the developmental abnormalities of mice with expression defects in Rasgrf1 or Igf256,57. Together, these findings suggest that the imprinted gene function based on differential expression pattern rather than parent-of-origin-specific manners. Importantly, these data support the assumption of “Conflict hypothesis”, which posits that genomic imprinting in mammals evolved to control the total expression levels of imprinted genes, with no qualitative differences between parental alleles58. However, how the current parental-origin pattern of the individual DMR is formed during evolution is an intriguing question59,60,61, which needs to be addressed in the future and may lead to the discovery of the origin of genomic imprinting.
Nevertheless, there are certain limitations in our Ha-Ha-fusion system. (1) Although 4N embryo complementation technology is a well-accepted strategy to test the in vivo developmental potential of injected pluripotent stem cells, the overall development potential of reconstructed embryos is limited due to the technical difficulties in ESC preparation and injection. We address this by using 4-cell 4N embryos instead of regular blastocysts as recipients, which improves the body chimeric proportion of fused ESCs and even germline transmission62,63. (2) Our strategy cannot analyze the role of imprinting in placental development. Nuclear transfer technology could address this limitation, but preliminary attempts showed that cloned embryos from XIK-DKO cells developed only to E10.5 (data not shown), in contrast to E13.5 of tetraploid complementation embryos with the same cells (Supplementary Fig. S2f). This result indicated that placental defects severely impaired the developmental potential of cloned embryos, which is consistent with our previous observation that defects in the trophoblast cell lineage account for the impaired in vivo development of cloned embryos21. In addition to DMR-regulated imprinted genes critical for placental development22,64,65,66,67,68,69,70,71, loss of H3K27me3 imprinting also disrupts post-implantation development of cloned embryos72. Rescuing the expression of H3K27me3-imprinted genes prevents the placental overgrowth in cloned animals73,74, highlighting the need to normalize H3K27me3-imprinted patterns for successful development of cloned embryos from Ha-Ha-fusion-derived cells. (3) CRISPR-Cas9-mediated DMR deletions may cause potential side effects, such as off-targets and even changes to the genome structure. To mitigate this, we analyzed at least 2 cell lines for each genotype and selected the cell lines with the best developmental potential for further analyses (Table 1). We also adopted the deletion strategy reported by previous references, in which the deleted DMRs have been proven to have the capacity to correct the monoallelic gene expression of the major genes in the imprinted cluster. Nevertheless, another strategy based on dCas9-mediated methylation rewriting reported by Wei et al. can avoid DNA deletions75, and thus might be very useful for future studies combined with the Ha-Ha-fusion system. (4) 8KO-DKO embryos develop to term efficiently. However, none of them survive to adult. This may result from DMR deletions failing to fully replicate physiological regulatory patterns (Fig. 3l, m; Supplementary Fig. S3g, h)44, or the absence of other critical imprinting loci in the Ha-Ha-fusion system. To address this, it is better to use haESCs from another genetic background such as JF1, which, based on SNPs, allows measuring the allele-specific expression of every imprinted gene in the genome after the different rescue attempts. This application can help us to better understand the changes in gene expression patterns after DMR deletions and probably reveal important clues that may be underlined in the lethality of 8KO-DKO embryos. Furthermore, this strategy can avoid the introduction of base mutations in the experiments of the DMR-switch between the paternal and maternal genome (Figs. 4h and 5f). In conclusion, while the Ha-Ha-fusion system provides a powerful platform for simultaneous manipulations of maternal and paternal imprints, further refinements are needed to enhance its efficiency and enable deeper exploration of imprinting network during development.
Materials and methods
Experimental mice
All animal procedures were performed under the ethical guidelines of Center for Excellence in Molecular and Cell Science (CEMCS), Chinese Academy of Sciences. All mice were housed in specific pathogen-free facilities of CEMCS. Female mice of BDF1 (C57BL/6×DBA/2) and ICR backgrounds were used to provide oocytes for micromanipulation and as pseudopregnant surrogates, respectively. CAG-RFP transgenic male mice (FVB/NJ background) were used to provide sperm for the establishment of androgenetic haploid ESCs. Actin-EGFP transgenic female mice (C57BL/6 background) were used to give oocytes for the establishment of parthenogenetic haploid ESCs.
Derivation of haploid cell lines
Derivation of PG-haESCs was performed as previously described19. To derive GFP-labeled PG-haESCs, Actin-EGFP transgenic female mice (8–10 weeks) were induced to superovulate. The GFP-labeled oocytes were cultured in CZB medium for 1 h and then activated for 5–6 h in an activation medium containing 10 mM Sr2+. Following activation, embryos were cultured in KSOM medium with amino acids in an incubator with 5% CO2 at 37 °C. Similarly, the derivation of AG-haESCs was carried out, as previously described12. Embryos were collected at blastocyst stage and were transferred into ESC medium supplemented with 15% FBS, 1000 U/mL LIF, 3 M CHIR99021 (Stemcell Technologies, 100-1042), and 1 M PD0325901 (Stemcell Technologies, 100-0248). To sort haploid cells, ESCs were trypsinized and incubated with 15 mg/mL Hoechst 33342 in a 37 °C water bath. Subsequently, the haploid 1 C peak was purified using BD FACS AriaII for further culturing.
CRISPR-Cas9-mediated imprinted region deletion in haploid cells
CRISPR-Cas9-mediated gene editing was performed as previously described18. To delete imprinted regions in haploid cells, sgRNAs were designed by sgRNA online design platform Chopchop (https://chopchop.cbu.uib.no) (Supplementary Table S1). Oligo DNAs were synthesized, and ligated to the pX330-mCherry (for GFP-PG-haESCs) or pX458-GFP (for RFP-AG-haESCs) plasmid (Addgene, 98750 and 48138). Plasmids were transfected into haploid ESCs by Lipofectamine 3000 (Invitrogen, L3000015). One day after transfection, the androgenetic and parthenogenetic haploid cells, respectively, expressing GFP and mCherry fluorescence protein were enriched by flow cytometry and plated at low density. Six days after plating, single colonies were picked for the derivation of haESCs. DMRs-deleted haploid cell lines were identified by DNA sequencing of PCR products amplified from targeted sites.
Genotyping of ESC lines and reconstructed pups
Genotyping of ESC lines and reconstructed pups was performed by Sanger sequencing of PCR products amplified from targeted sites and examination using fluorescence microscopy. For DNA sequencing, two pair primers were designed to check deleted regions and WT regions respectively (Supplementary Table S2). After double-checking by gel electrophoresis, PCR products were performed by Sanger sequencing. For examination with fluorescence microscopy, cultured cells and tails of pups were examined by FITC and PE-Cy5 channels of Olympus IX 51 microscopy.
Fusion of haploid cells to derive diploid cells
The AG- and PG-haploid cells, respectively, were suspended with 0.05% trypsin/EDTA and suspended in a fusion solution. Then two kinds of haploid cells were mixed and added with the Sendai virus (GenomONE, CF016EX). Mixture was incubated at 4 °C for 5 min and 37 °C for 15 min. Then mixture was plated in 6-well plate. After 48 h of plating, the diploid double positive (GFP and RFP) cells were purified by flow cytometry and plated at low density. 6 days after plating, single colonies were picked for the derivation of the diploid fused cells. The passage number of fused ESCs is inherited from the passage number of PG-haESCs.
Derivation of tetraploid embryos
To generate fertilization-derived tetraploid embryos, B6D2F1 females were superovulated and mated with B6D2F1 males. Zygotes were collected from the oviduct 18–24 h after hCG injection. Two-cell embryos were obtained at 1.5 dpc. To generate tetraploid embryos, electrofusion was performed on two-cell embryos to produce one-cell tetraploid embryos. Two-cell embryos were aligned using an alternating current in 0.3 M mannitol solution, a single alternating current pulse of 6 V/cm was applied for 5 s, followed by a direct current pulse of 1000 V/cm for 20 µs. After electrofusion, the embryos were washed and cultured in KSOM.
Tetraploid embryo complementation of ESCs
To generate tetraploid-complemented embryos, fused ES cells were fed with fresh medium 24–36 h before they were harvested for injection. Before injecting, ES cells were trypsinized, and suspended in HEPES-CZB medium. Previous studies have shown that 4-cell embryos could help ESCs exhibit high-proportion germline transmission62,63. Ten ESCs were microinjected into tetraploid 4-cell embryos to produce tetraploid-complemented embryos by blunt piezo-driven pipette. After culturing for one day, manipulated blastocysts were transferred into the uteri of the 2.5 dpc pseudopregnant mother. Recipient mothers carrying reconstructed embryos were euthanized at 19.5 dpc, and the pups were quickly removed from the uteri.
Intracytoplasmic AG-haESC or sperm injection and embryo transfer
To generate SC embryos, AG-haESCs were treated with 0.05 μg/mL Demecolcine solution (Sigma) for 10–12 h and synchronized to the M phase. Next, the M-phase clones were treated with trypsin and suspended in the HCZB medium. MII oocytes were collected from oviducts of superovulated B6D2F1 females (8 weeks old). AGhaESCs were injected into the cytoplasm of MII oocytes in a droplet of HCZB medium containing 5 μg/mL cytochalasin B (Sigma, C6762) using a Piezo-drill micromanipulator. The injected oocytes were cultured in the CZB medium for 30 min and then activated for 5–6 h in Ca2+-free CZB with SrCl2. For sperm injection, the procedures were the same as described above. Following activation, the reconstructed embryos were cultured in AA-KSOM (Merk, MR-106) medium at 37 °C under 5% CO2 in the air. The embryos obtained through Intracytoplasmic AG-haESC Injection (ICAHCI) were cultured in KSOM medium for 24 h to reach the two-cell stage. Thereafter, 18–20 two-cell embryos were transferred into each oviduct of pseudopregnant ICR female mice at 0.5 dpc. Recipient mothers carrying SC embryos gave birth at 19.5 dpc.
Bisulfite PCR
The genome extraction from ESCs used the TIANamp Genomic DNA Kit (TIANGEN, DP304-02). RNA was removed using RNaseA (ThermoFisher Scientific, R1253) in a 37 °C water bath for 1 h. DNA was recovered using a Universal DNA Purification Kit (TIANGEN, DP214-03). The bisulfite conversion was performed using the EZ DNA Methylation-Gold TM Kit (ZYMO research, D5005) for 200–500 ng genomic DNA, following the manufacturer’s instructions. The bisulfite DNA products were amplified by nested PCR. The amplified products were purified by gel electrophoresis using a Universal DNA Purification Kit and cloned into pMD™19-T Vector Cloning Kit (Takara, 6013). For each sample, more than 12 E. coli clones were picked for sequencing. The results were analyzed by the DNA methylation analysis platform. (http://services.ibc.unistuttgart.de/BDPC/BISMA/).
Histological analysis
For histological analysis, adipose tissues from newborn pups were fixed in formaldehyde solution. Then the samples were dehydrated and embedded in paraffin. Sections were prepared with an ultramicrotome (Leica, RM 2235). Serial 5-µm sections were cut and collected on glass slides. The sections were stained with H&E after drying and photographed with microscopy (IX 51, Olympus).
RNA extraction and real-time quantitative PCR (qPCR)
Total RNA was isolated from the cells using TRIzol reagent (Invitrogen, 15596018CN). The cDNA was obtained from about 1 μg RNA with a reverse transcription reaction by the HiScript III RT SuperMix for qPCR (Vazyme, R323-01). Real-time qPCR reactions (RT-qPCR) were performed on a Bio-Rad CFX96 using the ChamQ Universal SYBR qPCR Master Mix (Vazyme, Q711-02) in triplicate. All the gene expression levels were normalized to the internal standard gene Gapdh.
WGBS
A total amount of 5–10 μg genomic DNA was mixed with 25 ng lambda DNA and sonicated to 200–500 bp, followed by end repair and dATP addition using the homemade Kit. Next, methylated adapters (synthesized by ThermoFisher Scientific) were ligated to the sonicated DNA. AMPure XP Beads (Vazyme, N411) were used to remove < 200 bp fragments. Bisulfite treatment was performed using the EZ DNA Methylation-Gold TM Kit (ZYMO research, D5005). After bisulfite conversion, the single-stranded, uracil-containing DNA was subjected to 10–12 cycles of PCR reaction with Illumina TruSeq PCR primers and 2.5 U of Pfu Turbo Cx Hotstart DNA polymerase (Agilent Technologies, 600410) to recover enough DNA for sequencing. The sequencing reads were aligned to mm9 using BSMAP with parameters –r 0 –w 100 –v 0.1 –A AGATCGGAAGAGC. Multiple mapped reads and PCR duplicates were removed. After mapping, those reads with total CG coverage less than 5 within 200 bp were removed. The methylation level was calculated using methylated CpG versus total CpG in each bin. The file with DNA methylation levels for each 200 bp bin was used for further analysis. For the boxplots about DNA methylation, we calculated the average DNA methylation levels of each region for each kind of gene element. DMR site information is presented in Supplementary Table S3.
Responses