Horizontal metaproteomics and CAZymes analysis of lignocellulolytic microbial consortia selectively enriched from cow rumen and termite gut
Introduction
Lignocellulose (LC) is the major component of plant cell walls and the
primary source of renewable carbon on Earth available for a sustainable production
of biofuels and commodity chemicals. However, biomass bioconversion is very
challenging due to LC recalcitrance [1].
Therefore, to improve the economic feasibility of biomass biorefining, more
effective strategies are required to break down LC.
In natural ecosystems, LC decomposition is performed by complex
microbial communities producing large enzyme arsenals. These communities includes
the ones involved in the composting processes [2, 3], cellulose and
leaf-litter decomposition in soils [4,5,6], and the digestive systems of herbivores [7] and termites [8, 9]. In the case of
herbivores, LC decomposition is the result of the symbiosis between the animal host
and its digestive microbiome, the latter providing short-chain volatile fatty acids
(VFA) and microbial proteins to the host. Therefore, to improve the economic
feasibility of biomass biorefining, one strategy is to develop Nature-inspired
solutions by harnessing the power of naturally occurring LC-degrading microbiomes.
However, the challenge is to deploy such microbial ecosystems in controlled
bioreactor environments.
LC deconstruction is a complex process that marshals a large diversity
of enzymes rarely produced by a single microbial species [10]. The process involves carbohydrate-active
enzymes (CAZymes) that break down, modify or assemble glycans [11]. CAZymes are classified into 5 main
categories, namely glycoside hydrolases (GH), glycosyltransferases (GT),
carbohydrate esterases (CE), polysaccharide lyases (PL) and auxiliary activities
(AA), and their appended non-catalytic carbohydrate-binding modules (CBM)
[11]. Other non-catalytic protein
domains include cohesins (COH), dockerins (DOC) and S-layer homology (SLH) domains.
These are often appended to CAZymes and provide the means to incorporate cellulosome
complexes that link enzymes to the cell surfaces [12]. Microorganisms present in LC degrading environments such as
the digestive tract of termites or cow rumen provide vast reservoirs of CAZymes,
which are sources of new potent lignocellulolytic enzymes for biomass deconstruction
[13].
To better understand the biological mechanisms that underpin LC
degradation, meta-omics technologies are being used to investigate how microbial and
enzymatic synergies contribute to LC deconstruction [14]. 16 S rRNA gene amplicon sequencing and shotgun metagenomics
have provided information on both the taxonomic and metabolic potential of various
lignocellulolytic microbial communities [7, 10, 15]. Moreover, they have shed light on the
synergies between the members of these communities [2, 4, 16]. However, these approaches do not inform on
the actual metabolic activities of community members, nor do they provide details of
the effective role of genes in ecosystem functioning. For this, metaproteomics is a
precious tool since it provides information on the entire protein component of
microbial communities, links proteins to specific microbial taxa and correlates
their presence with metabolic activity [17]. This powerful approach thus provides useful information on
metabolic networks and symbiotic microbial interactions.
Previous metaproteomics studies on rumen ecosystems have shown that the
most abundant proteins are affiliated to Bacteroidetes (Bacteroidota), Firmicutes
(Bacillota) and Proteobacteria (Pseudomonadota) [18], where LC degradation is associated with high redundancy of
key enzyme activities. Regarding the termite gut microbiome, metaproteomics of
Nasutitermes corniger showed that among 197
identified proteins with known functions, 48 proteins are directly related to glycan
hydrolysis [19]. However, due to the
high complexity of these natural ecosystems and the limited number of proteins
detected, these metaproteomic studies neither revealed the specific roles of
individual microbial taxa, nor the temporal dynamics of proteins involved in LC
breakdown.
To gain insight into how lignocellulolytic ecosystems function,
selective ecosystem enrichment is often used to reduce microbial complexity and hone
community functions for use in bioprocesses [20,21,22,23].
However, most previous metaproteomic studies performed on simplified ecosystems only
revealed a small number of proteins [23,24,25,26,27]. They
also fail to fully capture the temporal dynamics of microbial species and CAZymes,
although one previous study has provided insight into the protein expression
dynamics of a subset of enzymes [28].
To further elucidate the mechanisms employed by microbial consortia to decompose LC,
it is thus vital to fully capture the temporal dynamics of all active species and
expressed proteins. Using LC as the sole carbon source, we postulate that
inoculum-specific microbial species can be maintained, although we expect that
time-dependent enzyme profiles to vary depending as a function of substrate
modifications occurring during the degradation process. Nevertheless, we also expect
all enzyme profiles will display genericity, regardless of the source of the
inoculum.
In previous studies, we reported the selective enrichment of two
anaerobic lignocellulolytic microbial consortia derived from cow rumen (RWS) and
from the termite gut microbiome of Nasutitermes
ephratae (TWS) [20,
21]. These naturally-occurring
anaerobic microbiomes present significant and contrasting levels of diversity, and
display great potential for LC degradation [7, 8, 13]. The enrichment of these microbiomes by a
sequential batch reactor process, using wheat straw as sole carbon source, resulted
in consortia displaying high LC-degradation activity and good ability to produce
carboxylates (mainly VFAs with acetate, butyrate and propionate as main products).
These products are valuable chemicals for producing bioplastics and liquid biofuels
[29, 30]. The kinetic characteristics of these enriched consortia were
determined by measuring LC-degradation rate, xylanase activity and carboxylate
production. Herein, we expand on previous work, performing shotgun metaproteomic
analysis over the reaction period of wheat straw hydrolysis by RWS and TWS
consortia.
Materials and methods
Lignocellulose degradation by RWS and TWS microbial consortia
The kinetic behaviors of the enriched lignocellulolytic microbial
consortia derived from cow rumen (RWS) and termite gut (TWS), summarized in
Table 1 and Figure S1, have been described previously
[20, 21]. For each consortium, two identical
anaerobic bioreactors were carried out, using a mineral medium containing wheat
straw as the sole carbon source (20 g.L−1).
Bioreactors were operated for 15 days at 35 °C under stirring (400 rpm) and pH
control (6.15), as detailed in Supplementary
Material and Methods.
The temporal dynamics of species and expressed proteins in RWS and
TWS along LC degradation were assessed by 16 S rRNA gene sequencing and shotgun
metaproteomics performed on four time points for each bioreactor. Time point
selection was based on wheat straw degradation, VFA production and xylanase
activity profiles (Table 1). The first
point (T1) corresponds to an early phase where xylanase activity and
lignocellulose degradation are low. Time points 2 and 3 (T2 and T3) correspond
with the start and end of maximal xylanase activity and peak lignocellulose
degradation rate. The fourth time point (T4) captures the final phase, when
wheat straw degradation, VFA production and xylanase activity stagnate.
Microbial diversity analysis
Total DNA/RNA were extracted from the pellet fraction of 1.5 mL
samples (13,000 x g, 5 min, 4 °C) using the
PowerMicrobiome isolation kit (Qiagen, Courtaboeuf, France) according to the
manufacturer’s instructions. After DNA purification (AllPrep DNA/RNA MiniKit,
Qiagen), the hypervariable V3-V4 region of the 16 S rRNA gene was amplified by
Illumina MiSeq sequencing (GenoToul Genomics and Transcriptomics platform,
Toulouse, France) using the conditions and primers previously described
[31]. Sequencing data was
analyzed using Find Rapidly OTUs with Galaxy solution (FROGS) pipeline
[32], as detailed
in Supplementary Information. R
CRAN software (v4.0.0) was used for further analysis. Diversity metrics were
obtained using R’s Phyloseq package v1.32.0 [33]. Sequencing data were deposited to the Sequence Read
Archive (SRA) under accession number PRJNA729464.
Metaproteomics analysis
Protein extraction, peptides digestion and mass spectrometry
analysis
Protein extraction was carried out on 3 technical replicates
per sample (3 mL), using a phenol buffer following the procedure for complex
sediment samples [34] and
separated by SDS-PAGE. After trypsin proteolysis (Promega, Fitchburg, WI,
USA) and purification of peptides (ZipTips, C18, Merck, Millipore,
Billerica, MA, USA), the dried samples were stored at −20 °C.
Mass spectrometry (MS) was performed on a Q Exactive HF MS
(Thermo Fisher Scientific, Waltham, MA, USA) with a TriVersa NanoMate
(Advion, Ltd., Harlow, UK) source in liquid chromatography chip coupling
mode. Peptide separation settings were as in procedures previously described
[35]. MS data have been
deposited into the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the proteomics identification (PRIDE) partner repository
[36]. Peptide
identification was performed with the Thermo Proteome Discoverer software
(v1.4; Thermo Fisher Scientific, Waltham, MA, USA) using Sequest HT against
bacterial sequences only (plant and eukaryotic sequences excluded) of
Uniprot-TrEMBL database (release date of 6th
April 2016) [37]. Peptide
identification considered a false discovery rate (FDR) below 1% calculated
by Percolator [38], a minimum
length of six amino acids, and a peptide rank of one. Protein matches were
only accepted if they were identified by a minimum of one unique peptide and
a high confidence. “Protein Groups” (hereafter referred to as proteins) were
form with strict parsimony and using the highest scoring protein in the
group as the confident master candidate protein. The detailed protocol is
included in Supplementary Material and
Methods.
Protein abundance, taxonomic and functional annotations
PROPHANE pipeline (www.prophane.de) [39] was used
for protein identification and taxonomic annotation using, respectively, the
highest sequence similarity to the UniProtKB/TrEMBL database and BlastP
considering Bacteria proteins only. Fungal proteins were not included as no
anaerobic fungi were detected by qPCR in RWS (data not shown) and no fungi
are present in the gut of higher termites [40]. Functional predictions of cluster of orthologous
groups proteins (COGs) and Pfam (Protein families) domains were obtained
with, respectively, RPS-BLAST algorithm and the COG collection (release
22.03.2003) [41], considering
the first hit for each protein (e-value ≤ 0.01), and the Hidden Markov Model
profiles with HMMER3 algorithm, considering the first hit for each protein
(gathering cut-off) [42]. Only
identified proteins were retained for further analysis. For each sample,
PROPHANE estimated protein abundance based on the normalized spectral
abundance factor (NSAF) [43].
Replicate-to-replicate variation was assessed by Pearson correlation
analysis using the cor R function, accepting a minimum value of 0.7.
Proteins present in only one technical replicate were discarded; remaining
proteins were expressed as mean values. Computational assignment of protein
functions were carefully checked, completed, and manually curated.
CAZymes annotation
The identified proteins were assigned to CAZyme families
following the day-to-day updates procedure of the CAZy database (accessed
March 30, 2018) as described previously [44, 45].
Briefly, protein sequences were compared to the full-length sequence of
previously annotated proteins, stored internally in the CAZy database, using
BlastP (version 2.3.0 + ). All remaining sequences with no hit were compared
with BlastP to a library of individual modules (catalytic or ancillary) and
a HMMER3 search against a collection of hidden Markov models based on each
CAZy family [45]. Presence of
signal peptides in CAZymes was predicted by SignalP 6.0 (https://services.healthtech.dtu.dk/services/SignalP-6.0/).
Data analysis and visualization
Hierarchical clustering (Ward method with Euclidean distance)
was used to group the samples with dist
and hclust R functions. Plots were
constructed using ggplot2 package v3.3.2. Relative abundances of bacterial
OTUs or proteins were aggregated at a target taxonomic level for stacked bar
plot representation. Low abundance taxa (<1% Phylum and <2%
Genus level), were gathered as “Other”. A unique protein affiliated to
Chitinispirillum, was excluded from
the analysis.
Statistical significance of differences was determined using
Wilcoxon tests with Benjamini-Hochberg p-value correction for pairwise
comparisons [46]. For
multivariate analysis, centered log-ratio (CLR) transformation of variables
[47] was performed to take
into account the compositional properties of our data. A correction factor
equivalent to 70% of the lowest value of each variable was applied to
eliminate zero values observed in the dataset [48, 49]. Microbial species and CAZymes that best discriminate
consortia and incubation time were investigated by principal component
analysis (PCA) and identified by multivariate integrative partial least
squares discriminant analysis (MINT-PLS-DA) performed with factoextra v1.0.7
and mixOmics v6.12.2 packages [50, 51].
Discriminant factors were validated with permutational multivariate analysis
of variance (PERMANOVA), using R’s vegan package v2.5–6 to test the
statistical significant differences [52].
Results
Data overview
16 S rRNA gene sequencing of the lignocellulolytic consortia RWS
and TWS generated 800,329 high quality reads (Supp. data 1); rarefying of samples to 15,000 sequences
captured most of the microbial diversity (Fig. S2). Metaproteomic analysis yielded a total of 10,342
proteins (Supp. data 1), with high
similarity between technical replicates (Pearson correlation >0.7;
Table S1) and similar coverage,
with an average of 2,792 proteins per sample (Table S2). 33.7% of total proteins were identified in both RWS and
TWS consortia (proteins with same accession numbers), while 35.4% and 30.9% of
proteins were specific to RWS and TWS, respectively. Hierarchical clustering of
overall proteins of RWS and TWS revealed consortia-specific profiles
(Fig. S3), evolving as a function
of LC degradation.
Taxonomic and functional profiles of RWS and TWS throughout wheat straw
degradation
Taxonomy deduced from 16 S rRNA gene sequencing data showed that
TWS displayed a lower richness and Shannon diversity index. However, the Shannon
diversity estimated from metaproteomics data [53] were very close for both consortia (Table S2). Indeed, a similar taxonomic composition
for the two consortia was revealed by the taxonomic affiliation derived from
both approaches, with a dominance of Bacteroidetes (Bacteroidota)(>
60%), Firmicutes (Bacillota) (about 20%) and Proteobacteria (Pseudomonadota)
(about 10%) phyla (Fig. S4). A minor
fraction of both communities belonged to Spirochaetes, while Fibrobacteres
(Bacillota) was exclusively found in RWS.
Affiliation of proteins at the genus level revealed that the active
populations, contributing most to protein production, belonged to Bacteroides in all samples of both consortia
(Fig. 1A), these remaining abundant
throughout the experiment. In second place was Clostridium, but its abundance decreased over time. Multivariate
PCA analysis of the abundance of all taxa-affiliated proteins, clearly
differentiated RWS and TWS communities (Fig. 1B). This difference was confirmed by a Wilcoxon test
(Fig. S5) showing a higher
abundance of proteins belonging to Dysgonomomas,
Fibrobacter and Enterococcus
in RWS while TWS showed a stronger abundance of proteins affiliated to Clostridium, Lachnoclostridium and Bacteroides, and to minor genera belonging to
Firmicutes-Others and Proteobacteria-Others. The temporal dynamics of the main
taxa of both consortia was evidenced by MINT-PLS-DA analysis, using the
incubation time as the discriminant factor. This revealed three clusters formed
by T1, T2/T3 and T4 samples (Fig. 1C).
T1 samples showed higher abundance of proteins affiliated to Clostridium, Shigella,
Enterococcus and Proteobacteria-Others (Fig. 1D) whose abundances decreased with incubation
time. Samples T2/T3 showed higher abundances of proteins affiliated to Lachnoclostridium, Prevotella and Firmicutes-Others while in samples T4, proteins
affiliated to Sphaerochaeta and Spirochaetes
showed higher abundance, which increased over the incubation period. Proteins
affiliated to Ruminococcus and Butyrivibrio were also abundant in T4 samples, but
also in T2 and T3 samples. PERMANOVA analysis (Table S3) confirmed that inoculum source (R-squared value 0.6214)
and incubation time (R-squared value 0.2022) were the key factors explaining the
protein-taxonomic composition and dynamics for both consortia.
dynamics.
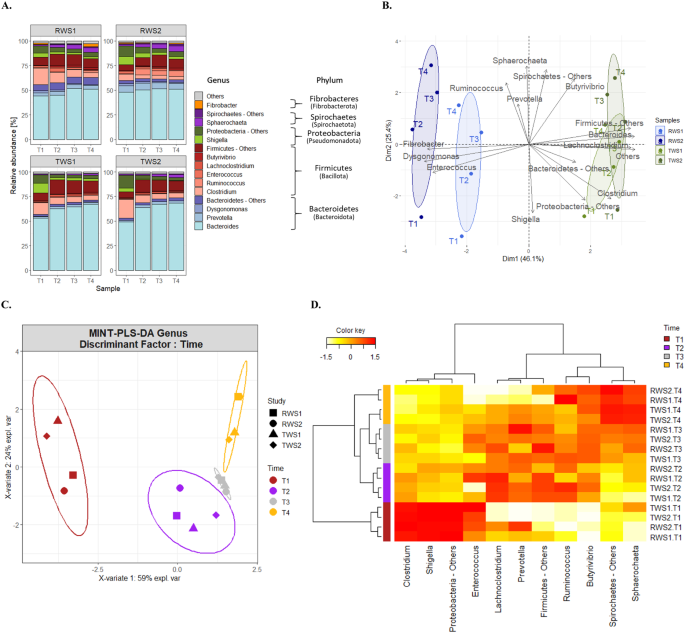
A Taxonomic community
composition at genus level deduced from the identified proteins
(metaproteomics data) for RWS (top graphs) and TWS (bottom
graphs) (1 and 2 indicate the biological duplicates). Relative
abundance of proteins based on NSAFs (normalized spectral
abundance factors) was aggregated at the genus level for stacked
bar plot representation. The group “Others” gather phyla with
relative abundance less than 1% in the dataset. Within major
phyla, the group “Others” gathers genera with relative abundance
less than 2% in the dataset or unclassified genera. Proteins
belonging to the same bacterial phylum were represented with the
same color palette: Bacteroidetes (Bacteroidota) (blue),
Firmicutes (Bacillota) (red); Proteobacteria (Pseudomonadota)
(green), Spirochaetes (Spirochaetota) (purple),
Fibrobacteres (Fibrobacterota) (orange). B Impact of the inoculum origin evidenced by
Principal components analysis (PCA) ordination; PCA component 1
and 2 explained respectively 46% and 25.4% of the total variance
and C. Impact of the incubation
time in the community dynamic of RWS and TWS metaproteomes,
assessed by Multivariate Integrative Partial Least Square
Discriminant Analysis (MINT-PLS-DA) based on the abundance of
taxa deduced from proteins (genus level, CLR-transformed data).
MINT-PLS-DA component 1 and 2 explained 59% and 24% of the total
variance. Ellipses at 95% confidence. D Clustered Image Map (CIM) represented the most
discriminant genera of the different sampling times for RWS and
TWS. CIM was built using the genera contributing the most to the
two first MINT-PLS-DA dimensions. Hierarchical clustering was
derived using the Euclidean distance and Ward methodology.
Genera are represented in columns and samples in rows. The boxes
on the left highlights the clusters discriminating the sampling
points (yellow, gray, purple and dark-red). The abundance of
proteins affiliated to each genus is indicated by the
white-to-red color gradient (increasing values).
The status of lignocellulose-related functions of RWS and TWS,
revealed by COGs prediction from the protein dataset, revealed that the cellular
function “metabolism” was highly represented (about 45%) (Fig. S6A), with a strong representation of the
subrole “carbohydrate transport and metabolism” (about 15%), irrespective of the
sampling time and consortia. Bacteroidetes (Bacteroidota) made the greatest
contribution to proteins involved in this function (Fig. S6B), while Firmicutes (Bacillota) and
Proteobacteria (Pseudomonadota) provided a lesser contribution to this function.
Nevertheless, the normalization of proteins abundance at the phylum level,
revealed that the specific COG profile of Firmicutes (Bacillota) and
Proteobacteria (Pseudomonadota) members was particularly focused on the
“carbohydrate transport and metabolism” function (Fig. S6C), whereas Bacteroidetes (Bacteroidota)
species were involved in a wider range of functions.
Carbohydrate-active enzymes in RWS and TWS consortia
RWS and TWS expressed a large diversity of CAZymes involved in
plant cell wall degradation, with 423 proteins containing at least one CAZyme
domain (Supp. data 2). In silico prediction of signal peptides showed 222
CAZymes with a signal peptide (Supplementary data 1). 174 CAZymes (41%), distributed in 69 families, where
common to both consortia (Fig. S7A),
accounting for about 80% of the CAZymes abundance while 127 and 122 proteins and
15 and 10 CAZy families were exclusively found in RWS or TWS, respectively. Most
of these proteins contained GH domains, representing more than 70% of the CAZyme
domains detected, followed by CE domains (12–20%), CBMs
(9.5–12.5%), GT domains (5–8%) and PL domains (about 2%). These
CAZyme domains showed a higher average abundance in TWS than in RWS
(Fig. S7B). The main purveyor of
CAZymes in both consortia were Bacteroidetes (Bacteroidota) members, producing
236 proteins corresponding to about 80% of the CAZyme abundance
(Fig. S7C) followed by Firmicutes
(Bacillota) (138 proteins, average 15%) while Proteobacteria (Pseudomonadota)
expressed a minor fraction of them (24 proteins, <1.5%).
The most highly represented CAZymes are typically active on
hemicellulose and mainly affiliated to Bacteroidetes (Bacteroidota)
(Fig. 2A), except those classified
GH11. These proteins are putative hemicellulases with endo-β−1,4-xylanase (GH8,
GH10, GH11) and β-mannanase (GH26) activities (Supplementary data 2). Hemicellulose-oligosaccharide-degrading
enzymes, with mainly β-xylosidase or α-L-arabinofuranosidase functions (GH43),
and hemicellulose-debranching enzymes with α-L-arabinofuranosidase (GH51) and
xylan α−1,2-glucuronidase (GH67, GH115) activities were also abundant.
Cellulose-degrading enzymes were mainly annotated as β-glucosidases (GH3) of
Bacteroidetes (Bacteroidota) origin, while those with endoglucanase activity
mainly belonged to Firmicutes (Bacillota)(GH5, GH9, GH48) (Fig. 2A). Some of these proteins, were appended to
CBMs or cellulosome (COH and DOC) domains. Among the most prevalent proteins in
both consortia were Bacteroidetes (Bacteroidota)- and Firmicutes (Bacillota)-
affiliated proteins related to starch degradation and including α-galactosidase,
1,4-α-glucan branching enzyme, α-glucosidase and α-amylase activities. According
to our data, Proteobacteria (Pseudomonadota) were only minor CAZymes
contributors, being mostly sources of acetyl-xylan esterase activity (CE1;
Fig. 2B). Non-catalytic carbohydrate
binding modules (CBM), SLH, COH and DOC (i.e., cellulosome components) were mainly Firmicutes (Bacillota)
origin (Fig. 2B).
RWS and TWS.
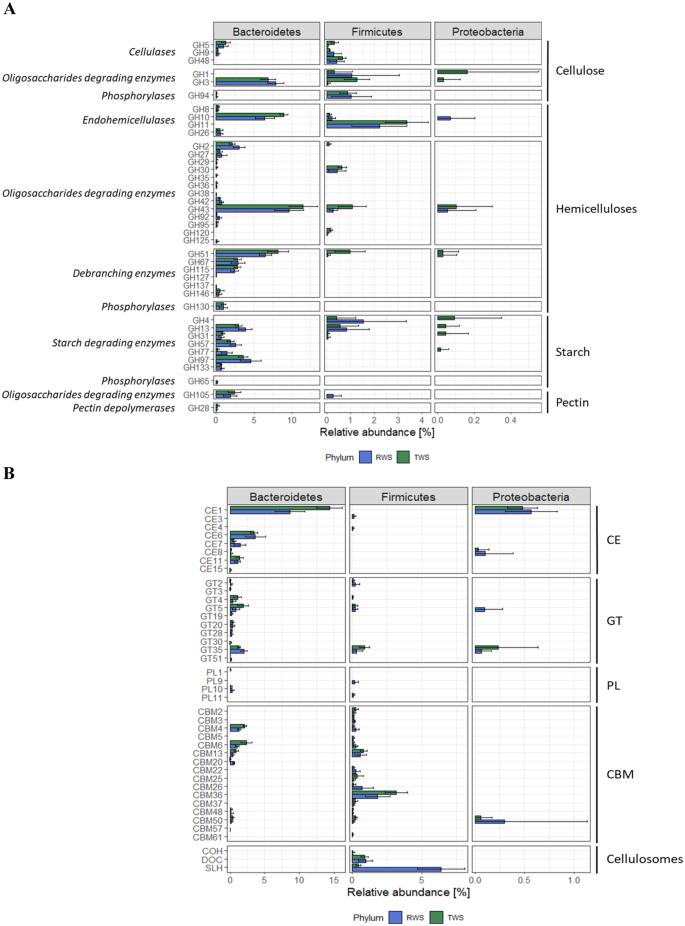
A Relative abundance of
glycoside hydrolases (GH) in Bacteroidetes (Bacteroidota),
Firmicutes (Bacillota) and Proteobacteria (Pseudomonadota) phyla
of RWS and TWS metaproteomes. Abundances were normalized to
total CAZymes expression. Only GH targeting cellulose,
hemicelluloses, starch and pectin fractions are showed. Error
bars represent the standard deviation. B Relative abundance of carbohydrate esterases
(CE), glycosyl transferases (GT), polysaccharide lyases (PL),
carbohydrate-binding modules (CBM) and cellulosomes domains
(S-layer homology (SLH), dockerins (DOC), cohesins (COH))
present in Bacteroidetes (Bacteroidota), Firmicutes (Bacillota)
and Proteobacteria (Pseudomonadota) phyla in RWS and TWS
metaproteomes. Abundances were normalized to total CAZymes
expression. Error bars represent the standard
deviation.
CAZymes involved in lignocellulose degradation
Multivariate statistical analyses were used to investigate how both
the inoculum origin and progression of LC degradation influenced the expressed
CAZyme profile. The PCA clearly separated RWS and TWS samples in function of the
inoculum (Fig. S8A, first component)
while the initial time points (T1) were clearly separated from the others
(Fig. S8B, second component).
PERMANOVA analysis (Table S4)
confirmed that the inoculum source and incubation time (R-square values of 0.253
and 0.284, respectively) were the key parameters determining the CAZyme
expression profiles. The importance of incubation time was confirmed by
Multigroup Supervised Partial Least Squares Discriminant Analysis (MINT-PLS-DA),
using the incubation time as the discriminant factor. This analysis separated
the initial point (T1) from the subsequent sampling points (T2/T3) and from the
final point (T4) (Fig. 3A). T1 samples
(Fig. 3B, dark blue box) clustered
CAZymes with oligosaccharide-degrading activities linked to cellulose (GH1) and
hemicelluloses (GH43), pectin methylesterase (CE8), and α-glucan degradation
(GH4, GH13, GH31); they also included starch-specific CBMs (CBM20, CBM25,
CBM26). Other CAZymes with hemicellulose-oligosaccharide degradation (GH2 and
GH29) or polysaccharide lyase (PL9) activities, were also found in T1 samples
(Fig. 3B, light-blue box). However,
this enzyme cluster was also present (at a lower abundance) at the end of the
incubation period (T4). The cluster formed by the sampling points T2 and T3
(Fig. 3B, purple box), where most of
the LC degradation occurred, was characterized by high abundance of
endoglucanases (GH5, GH9 and GH48) and a large panel of hemicellulolytic
CAZymes, including endo-hemicellulases (GH11), deacetylating (CE1, CE11) and
debranching (GH67, GH115) enzymes and enzymes active on
hemicellulose-oligosaccharides (GH30, GH27). CAZymes related to
pectin-oligosaccharide degradation (GH105) were also found, as well as xylan- or
amorphous cellulose-specific CBMs (CBM3, CBM4, CBM13, CBM22, CBM36), dockerins
typical of cellulosome structures, and glucosyl/galactosyltransferases (GT4,
GT5). This cluster of CAZymes was also highly abundant at the latter stage of LC
degradation (T4; Fig. 3B, dark-green
box) along with a range of enzymes including endo-hemicellulases (GH10) and
xylan deacetylating (CE6) or hemicellulose-oligosaccharides degrading enzymes
(GH36, GH125) (Fig.3B, light-green box).
CBMs specific for cellulose (CBM30), hemicellulose (CBM11) and starch (CBM48),
and starch phosphorylases (GT20, GT28) completing this cluster. Similar results
were observed when the analysis was performed for CAZymes with predicted signal
peptide only (Fig. S9).
metaproteomes.
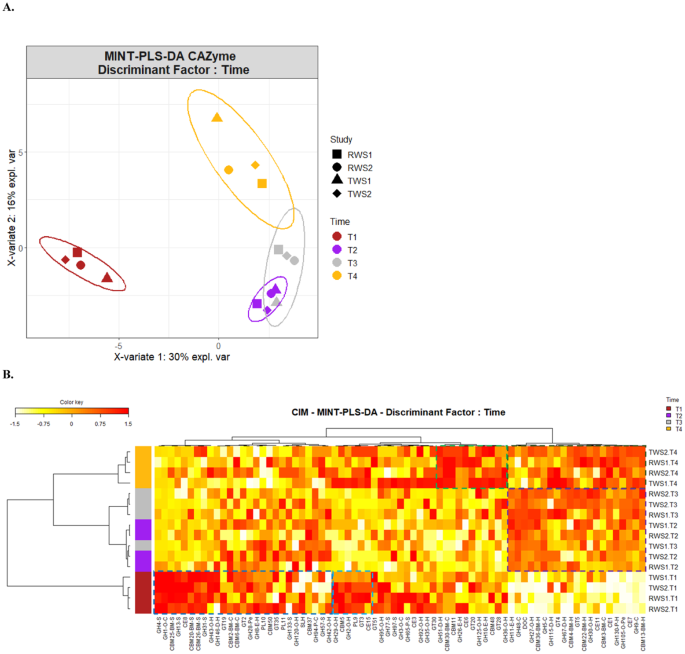
A Impact of the
incubation time in the CAZyme dynamics of RWS and TWS
metaproteomes, assessed by Multivariate Integrative Partial
Least Square Discriminant Analysis (MINT-PLS-DA) based on CAZyme
abundances (family level, CLR-transformed data). MINT-PLS-DA
component 1 and 2 explained 30% and 16% of the total variance.
Ellipses at 95% confidence. B
Clustered Image Map (CIM) represented the most discriminant
CAZyme families of the different sampling times in RWS and TWS.
CIM was built using the main CAZyme families explaining the
first two MINT-PLS-DA dimensions. Hierarchical clustering
(Euclidean distance and Ward method) represents CAZymes in
columns and samples (T1–T4) in rows. The boxes on the
left highlights the clusters discriminating the sampling points
(yellow, gray, purple and dark-red). The abundance of CAZy
families is indicated by the white-to-red color gradient
(increasing values).
Enzymes related to volatile fatty acid production in RWS and TWS
consortia
Lignocellulose degradation by RWS and TWS, like in other microbial
consortia, is associated with VFA production [40, 54]. RWS and
TWS produced mainly acetate, propionate and butyrate with a higher proportion of
the latter in TWS (Table 1; average
molar ratio of acetate:propionate:butyrate of 59:23:18 for RWS and 47:20:33 for
TWS). Based on previous COG assignments [16, 55,
56], metaproteomic data
revealed that the key enzymes involved in VFA biosynthesis constituted
2.8 ± 0.4% and 3.5 ± 0.2% of total protein abundance in RWS and TWS,
respectively. According to the higher butyrate production measured in TWS,
proteins related to butyrate biosynthesis (219 proteins) were more abundant in
this consortium (Figure S8). In both
consortia, this function was associated with a large variety of enzymes (numbers
of proteins indicated in parenthesis): acetyl/propionyl CoA carboxylase (3),
butyrate kinase (7), 3-hydroxyacyl-CoA dehydrogenase (26), enoyl-CoA hydratase
(17), acetyl-CoA acetyltransferase (52), alcohol dehydrogenases YqhD (8) and
class IV (67), short-chain alcohol dehydrogenase (38) and Zn-dependent alcohol
dehydrogenase (1). Most of them were affiliated to Firmicutes (Bacillota) (138
proteins) and Bacteroidetes (Bacteroidota) (23 proteins), but with abundance
levels similar for both phyla (Fig. 4).
At the initial phase of incubation (T1), Clostridium species were the major contributors of butyrate
biosynthesis enzymes in both consortia (67 proteins, about 30% abundance), but
subsequently the abundance of proteins affiliated to Bacteroides (16 proteins) increased, with higher levels of
expression being reached in the latter phase of biomass bioconversion.
VFAs production.
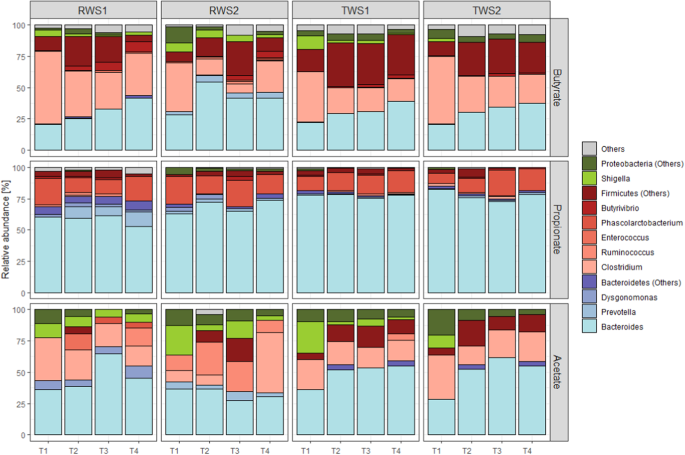
Relative abundance and phylogenetic origin of bacterial
enzymes involved in butyrate, propionate and acetate production
in the metaproteomes of RWS and TWS (1 and 2 indicate the
biological duplicates). Taxonomic affiliation of proteins at the
genus level. Proteins belonging to the same bacterial phylum
were represented with the same color palette: Bacteroidetes
(Bacteroidota) (blue), Firmicutes (Bacillota)(red) and
Proteobacteria (Pseudomonadota) (green). For butyrate
biosynthesis: COG4770 (acetyl/propionyl-CoA carboxylase),
COG3426 (butyrate kinase), COG1250 (3-hydroxyacyl-CoA
dehydrogenase), COG1024 (enoyl-CoA hydratase), COG0183
(acetyl/butyryl-CoA acetyltransferase), COG1979 (alcohol
dehydrogenase YqhD), COG1454 (alcohol dehydrogenase, class IV),
COG1028 (short-chain alcohol dehydrogenase), COG1064
(Zn-dependent alcohol dehydrogenase). For propionate
biosynthesis: COG4799/0777 (acetyl/propionyl-CoA carboxylase),
COG2185/COG1884 (methylmalonyl-CoA mutase), COG0346
(methylmalonyl-CoA epimerase). For acetate production: COG1012
(NAD-dependent aldehyde dehydrogenase), COG0282 (acetate
kinase).
Propionate production was similar in both consortia
(Table 1) and appeared to be
associated with the presence of 66 propionate biosynthesis-related proteins with
three main functions: acetyl/propionyl-CoA carboxylase (34 proteins),
methylmalonyl-CoA mutase (23 proteins) and epimerase (9 proteins), revealing
that propionate was formed via the succinate pathway in both consortia. These
proteins were mainly affiliated to Bacteroidetes (Bacteroidota), particularly to
Bacteroides (37 from 66 proteins)
(Fig. 4), while in Firmicutes
(Bacillota), Phascolarctobacterium was the
main contributor to this activity, providing about 20% of the relevant
enzymes.
Surprisingly, for both consortia and irrespective of the sampling
time, proteins involved in acetate biosynthesis were the least abundant among
the VFA-biosynthesis enzymes (61 proteins accounting for about 0.3% of the total
protein abundance; Fig. S10). The
presence of acetate kinases (26 proteins), phosphate acetyltransferases (22
proteins), and aldehyde dehydrogenases (13 proteins), expressed by all phyla
(Fig. 4), suggests that acetate
production in the consortia occurrs via the Wood-Ljungdahl pathway. Seven of
these proteins affiliated to Bacteroides
formed the most abundant group, representing 39.3% and 49.3% of enzymes involved
in acetate biosynthesis in RWS and TWS metaproteomes, respectively, while those
belonging to Clostridium accounted for about
20% (Fig. 4). A low abundance of
proteins involved in carboxylate biosynthesis and belonging to Proteobacteria
(Pseudomonadota) was also detected, in particular for acetate and butyrate
production, at early stages of incubation.
Discussion
To gain new knowledge pertaining to LC-degradation process mediated by
microbial consortia, we studied two lignocellulolytic consortia, RWS and TWS,
derived from the complex microbiomes of the cow rumen and termite gut, respectively.
Compared to previous investigations, metaproteomic analysis of these consortia
enabled the detection of a high number of non-redundant proteins (10,342)
[23, 25, 28, 57], providing a complete repertoire of enzymes
involved in LC degradation and VFA production.
Although RWS and TWS are derived from contrasting parental microbiomes,
the taxonomic community composition deduced from 16 S rRNA gene sequencing and
metaproteomic data showed that the active communities of both consortia were rather
similar. They displayed a prevalence of Bacteroidetes (Bacteroidota)- and Firmicutes
(Bacillota)-affiliated proteins, with the genera Bacteroides and Clostridium being
the main representatives of these phyla, respectively. This similarity is probably
the result of the selective pressure exerted by the wheat straw substrate during the
enrichment process, as reported in previous studies [58,59,60].
Bacteroides and Clostridium are key players in LC degradation in the digestive
microbiomes of herbivores, where they break down complex carbohydrates particularly
cellulose, xylan and starch [61,
62]. 16 S rRNA and metaproteomic
data confirmed that the most abundant genera were also the major source of plant
cell wall degrading enzymes in RWS and TWS consortia, in agreement with previous
observations [18]. This nevertheless
contrasts with previous metatranscriptomics studies on the termite gut [63] and studies performed on enriched microbial
consortium able to degrade rice and wheat straw [27], sugar cane bagasse [23] and a corn-stover degrading consortium [28]. Our findings reveal that the relationship
between the abundance of microbial phyla and their functional impact on the
community activity is not a simple one, and highlights the importance of
metaproteomics to assign functional roles to different phyla.
The metabolic functions of RWS and TWS, assessed by COG analysis,
revealed that proteins related to “translation, ribosomal structure and biogenesis”,
“carbohydrate transport and metabolism” and “energy production and conversion” were
dominant in both communities, as reported in previous studies [16, 28, 57]. These COG
functions were expressed by the three main phyla found in RWS and TWS, Bacteroidetes
(Bacteroidota), Firmicutes (Bacillota) and Proteobacteria (Pseudomonadota), but
phylum-normalized data showed that protein expression by the two latter was strongly
directed towards carbohydrate metabolisms, while the former (the major phylum
Bacteroidetes – Bacteroidota) was associated with larger spectrum of
functions.
A large diversity of CAZymes related to LC degradation, including
cellulases, hemicellulases and CBMs were detected in RWS and TWS metaproteomes. They
belong to 94 families, illustrating the relative richness of these consortia when
compared to the whole CAZy database (n = 472).
CAZymes in RWS and TWS accounted for about 4% of total proteins expressed. Although
most previous studies report the number of non-redundant CAZymes, rather than their
abundance [16, 18, 28], the abundance reported here is comparable to that measured
(3.5% and 5.9%) for other lignocellulolytic consortia [27]. This implies that dedicating less than 5%
of their expressed proteins to LC degradation, RWS and TWS achieved high
(>45% in our study) biomass degradation levels.
Taxonomic affiliation of CAZymes revealed that LC degradation by RWS
and TWS results from the combined action of proteins affiliated to Bacteroidetes
(Bacteroidota), the main purveyors of CAZymes, followed by Firmicutes (Bacillota). A
closer inspection revealed that the Bacteroidetes (Bacteroidota) members mainly
produce hemicellulose-debranching and oligosaccharide-degrading enzymes as well as
enzymes involved in starch degradation. In contrast, Firmicutes (Bacillota) members
produced enzymes specific for β-glucan and β-xylan (cellulases and hemicellulases).
This differential expression of CAZymes offers interesting prospects for engineering
synergy. Moreover, it earmarks these two phyla as the key players in wheat straw
degradation. It is noteworthy that other studies do not systematically identify
these phyla as the dominant purveyors of CAZymes, particularly when considering the
termite gut microbiome [64]. To
rationalize this observation, we suggest that our results are the consequence of the
combined contributions of the original microbial consortium and the specific process
conditions employed [23, 26,27,28]. The
substrate, the enrichment process and the availability of oxygen all determine the
relative development of obligate aerobes, facultative aerobes and strict
anaerobes.
Although RWS and TWS consortia displayed taxonomic similarities, only a
third of proteins were common to both consortia. Multivariate analysis highlighted
the taxa and proteins linked to the parental inoculum source. Indeed, some features
can be attributed to the initial cow rumen and termite gut microbiome. For example,
the larger diversity of CAZymes and the ruminococcal and clostridial cellulosome
components characteristic of RWS are also characteristic of the cow microbiome
[65, 66]. Similarly, the high abundance of GH10, GH43, CE1 and
especially GH11, typical of TWS, is also a feature of termite gut microbiome
[67, 68].
Despite differences between RWS and TWS, multi-group MINT-PLS-DA
supervised analysis highlighted in both consortia Bacteroidetes (Bacteroidota)- and
Firmicutes (Bacillota)-affiliated enzymes, particularly ones produced by Bacteroides, Clostridium and Enterococcus genera, were marshalled. According to annotation, these
enzymes target the minor starch fraction and holocellulose-derived oligosaccharides,
meaning that the aforementioned genera forage ready available resources and in doing
so facilitate further breakdown of holocellulose polymers. Similarly, in both
consortia the occurrence of Lachnoclostridium,
Prevotella and Other-Firmicute members was
associated with the principal LC biomass degradation phase, which was also related
with an increase in the abundance of CAZy families related to cellulose hydrolysis,
deacetylation and cleavage of hemicelluloses and pectin depolymerization. In
addition, during the last incubation stage, several CAZymes hydrolyzing and
deconstructing hemicellulose and hemicellulose-oligosaccharides also increased, as
well as various CBMs binding cellulose, hemicellulose and starch. Therefore, further
biochemical characterization of these enzymes could be of interest to elucidate
their specific role in LC-biomass degradation, in particular to identify enzymes
able to degrade the most recalcitrant components.
Concerning VFA biosynthesis, our data showed that acetate biosynthesis
was the result of Wood-Ljungdahl pathway in both our consortia, and highlighted the
major role of acetogenic bacteria belonging to Bacteroidetes (Bacteroidota) and
Firmicutes (Bacillota) phyla. Propionate production in RWS and TWS mostly resulted
from the succinate pathway as evidenced by the detection of methylmalonyl CoA
mutases and epimerases [69], with
Bacteroides and Phascolarctobacterium species as the key propionate producers. This
is consistent with previous data that showed that the succinate pathway is the main
route reported for rumen [70]. Butyrate
biosynthesis resulted either from the conversion of butyryl-CoA into butyrate, using
butyrate kinase (synthesizes butyryl-phosphate) and phosphotransbutyrylase, or the
transfer of coenzyme A (catalyzed by butyryl-CoA:acetate-CoA transferase) between
acetate and butyrate. Although both routes are exploited by Firmicutes (Bacillota)
species, the second one was strongly enhanced in both consortia. This observation is
consistent with previous studies on the cow rumen and human gut microbiota that
revealed that Bacteroidetes (Bacteroidota) are responsible for the majority of
acetate and propionate production, while butyrate biosynthesis is mainly handled by
Firmicutes (Bacillota) species [16,
71]. LC-hydrolysis remains the
limiting step and thus VFA-biosynthesis cannot be used to improve LC conversion into
VFA, but could be of interest to drive VFA production towards specific products
(e.g. butyrate).
A remarkable finding in this work is that no lignin-specific enzymes
(CAZyme AA class) were found, suggesting that ligninolysis did not occur under the
anaerobic culture conditions used. This concurs with previous lignin measurements
performed on RWS and TWS [20,
21]. Nevertheless, the fact that no
lignin-degrading enzymes were evidenced does not imply that these were absent,
because shotgun metaproteomics procures an incomplete image of expressed proteins.
Ultimately, to assert that no lignin degradation had occurred in our experiments, it
would be necessary to perform a thorough physicochemical and structural analysis of
the substrate before and after microbial treatment.
The CAZy classification database has been growing at a fast rate in
recent years, with new sequences being added daily and new families being regularly
defined. A comparison of the collection of GHs detected in our study with those
detected in previous omics studies (metatranscriptomics, metaproteomics,
metagenomics) performed on bovine rumen and termite-gut microbiomes
(Table S5 [65, 72,73,74,75,76,77,78,79])
revealed that this study unmasked greater diversity of cell wall degrading CAZymes.
Moreover, the comparison revealed that our study captured most CAZy families
degrading the plant cell wall, whereas the previous studies were less successful in
this regard. To a large extent, the greater coverage of GH families in our study can
be correlated with the growth of the CAZy database and its date of access
[11] and ongoing improvements to
the experimental techniques and bioinformatics pipelines used. However, we believe
it is also attributable to the fact that RWS and TWS are enriched consortia whose
functions are highly adapted for the degradation of raw lignocellulosic biomass.
Undoubtedly, future studies benefitting from further progress in metaproteomics and
the further expansion of the CAZy database will surpass our study. Hopefully, these
will provide an even deeper understanding of lignocellulolytic functions in
microbial ecosystems and provide the means to identify proteins that are currently
unclassified. Furthermore, our data showed that RWS and TWS consortia represent
excellent simplified models to study the mechanisms governing the complex
lignocellulose degradation process and to better understand and exploit multispecies
lignocellulolytic enzyme systems for biotechnological applications.
Responses