Implication of CXCR2-Src axis in the angiogenic and osteogenic effects of FP-TEB

Introduction
Addressing large segmental bone defects (LSBD) remains a substantial clinical challenge. Tissue engineered bones (TEBs), involving the integration of viable osteogenic progenitors, typically mesenchymal stem cells (MSCs), and biocompatible scaffolds, hold significant promise for therapeutic applications1,2. However, the widespread clinical adoption of TEBs faces numerous obstacles originated from their inherent reliance on viable cells. It is of great significance to develop alternative biomaterials that are easier to prepare, store, transport, and more cost-effective.
Accumulating evidence indicates that MSCs implanted alongside scaffolds become largely undetectable within a short time frame, typically no more than 30 days. Most newly-formed bones appears to originate from host cells3,4. These phenomena have sparked discussions on the trophic effects of MSCs. Recent literature suggest that MSCs primarily create a favorable microenvironment for tissue regeneration rather than directly contributing osteogenic cells5. Spontaneously, MSCs secrete a diverse array of bioactive factors capable of attracting host cells and stimulating adjacent cells to participate in tissue repair6. Furthermore, their extracellular matrix (ECM) are well established to be implicated in tissue regeneration as they share similar bio-characteristics with resident cells in terms of inflammation regulation, immunity modulation, vasculature development and tissue repair5,7,8. In this context, we have previously introduced a novel biomaterial termed functional protein-based TEBs (FP-TEBs), developed through systematic freeze-drying of TEBs to devitalize MSCs while preserving most functional proteins9. FP-TEBs have exhibited marvelous osteogenic potential in vitro10. Given the close interplay between osteogenesis and angiogenesis, with adequate blood supply being a prerequisite for bone healing, it is hypothesized that FP-TEBs possess favorable angiogenic capacity due to retained proteins. However, FP-TEBs lack internal blood vessels or living cells prior to implantation, potentially resulting in incompetent vascularization due to local vessel destruction. Establishing information regarding the discrepancy between advantageous osteogenic potential and poor angiogenic environment is crucial for understanding the functional mechanism of FP-TEBs and guiding product innovation and development.
During the postnatal phase, the formation of new vascular networks in wound healing and tissue repair primarily relies on angiogenesis, a complex process involving the sprouting of endothelial cells (ECs) from pre-existing vessels11. The entry of ECs into the injury site is essential for initiating angiogenesis, encompassing EC proliferation, tube formation, and vascular remodeling12. Chemokines play critical roles in EC filtrations. In previous studies, we have primarily characterized the protein profile of FP-TEBs, revealing a significant presence of C-X-C motif ligands (CXCLs), particularly growth-regulated oncogenes, also known as CXCL1/2/3, CXCL5, CXCL6, and CXCL8. Intriguingly, these chemokines are engaged in various EC behaviors, such as migration, viability, apoptosis, and proliferation13,14,15. Their receptors, primarily C-X-C motif chemokine receptors (CXCRs) commonly constitutively expressed by ECs, are presumed to exert substantial influence on angiogenesis and osteogenesis16. Additionally, a distinct capillary subtype characterized by high expression of CD31 and Emcn, known as Type-H ECs, has been identified as pivotal mediators of angiogenesis and osteogenesis in bone tissues17. Subsequent investigations have underscored their crucial roles in guiding the directed migration of osteoprogenitor cells during bone repair. Consequently, elucidating the interplay between chemokines, Type-H ECs, angiogenesis, and osteogenesis holds significant potential for advancing the development and implementation of cell-free tissue engineering approaches18. In this context, this study sought to evaluate the in vivo angiogenic and osteogenic capacity of FP-TEBs and illustrate their functional way and underling mechanisms.
Results
FP-TEBs exhibit appreciable osteogenic and angiogenic capacities
During bone repair, angiogenesis, and osteogenesis are intricately linked, with angiogenesis appearing to precede osteogenesis due to the guidance provided by Type-H ECs in directing the migration of osteoprogenitor cells17. Consequently, the angiogenic capabilities of implants were primarily assessed, followed by the osteogenic abilities (Fig. 1a). By postoperative day 14, a greater number of newly formed blood vessels were observed within and surrounding TEBs and FP-TEBs, as compared to DBM (Fig. 1b). This finding aligned with existing literature demonstrating the ability of donor MSCs and their extracellular vesicles to enhance the vascularization of implants19. From the microscopic perspective, larger amounts of host CD31+ ECs were present in the zones of TEBs and FP-TEBs, as compared with DBM at day 14. Unexpectedly, a subdivision of ECs demonstrated that compared to TEBs, FP-TEBs showed superior attractive power on Type-H ECs (CD31hiEmcnhi), suggesting robust osteogenic potential therein (Fig. 1c). Consistently, the expression of RUNX2, an early osteogenic marker, was higher in FP-TEBs. Regarding ALP and OCN, the middle and late markers, respectively, no discernible distinction was observed between TEBs and FP-TEBs, despite that their expression levels were significantly higher than those of DBM (Fig. 1d). Immunofluorescence showed higher expression of OCN or COL1A1 in FP-TEBs and TEBs compared to DBM (Supplementary Fig. 1). The number of osteoclasts were consistently low in all three groups and no significant variance was observed among implants at 2 weeks postoperatively (Supplementary Fig. 2). We proceeded to evaluate the osteogenic potential of FP-TEBs. Micro-CT was applied and TEBs and FP-TEBs scaffolds showed more new bone formation than DBM. Quantitative analysis showed that bone tissue volume (BV), and number of trabecular bone (Tb.N) were significantly higher in the TEBs and FP-TEBs than DBM, while the trabecular space (Tb.Sp) showed the opposite pattern (Fig. 1e). At 4 weeks postoperatively, near-complete bony fusion was achieved within TEBs and FP-TEBs, with implants being encompassed by chondrocyte- and osteoblast-like cells and filled with viable osteocytes, signifying substantial bone regeneration. These findings implied that both of TEBs and FP-TEBs were capable of forming new bone tissues through endochondral ossification. In DBM scaffolds, few viable osteocytes were found in lacunas within bone pieces, and implants were poorly embedded by osteogenesis-related cells. Moreover, the number of blood sinuses was significantly greater within TEBs and FP-TEBs than DBM, suggesting advanced vascularization therein (Fig. 1f). At 8 weeks, the union rates of FP-TEBs and TEBs were comparable and both of them were significantly higher than that of DBM (Supplementary Fig. 3). Together, these findings demonstrated that TEBs and FP-TEBs exhibited comparable and appreciable capacities of bone repair. Meanwhile, early osteo-angiogenesis coupling in FP-TEBs seemed superior to that in TEBs, endowing the functional proteins retained in FP-TEBs with a fascinating glimpse.

a Schematic diagram of the experiments for assessing the osteogenic and angiogenic effects. b Representative Micro-fill perfusion images of bone defect areas from C57 mice treated with DBM, TEBs, and FP-TEBs at 2 weeks postoperatively. c Representative coimmunostaining images with quantification ofcell numbers in critical-size bone defects at 2 weeks postoperatively (n = 5). White arrows, staining-positive cells. Scale bar, 10 μm. d Comparison of mRNA and protein expression of osteogenic markers in critical-size bone defects at 2 weeks postoperatively (n = 3). e Micro-CT reconstruction and quantitative analysis of the bone volume (BV), Tb. Sp (Trabecular separation), and Tb. Th (Trabecular thickness) of bone defect areas at 4 weeks postoperatively (n = 5). Dotted straight red line indicates the area of interest for quantitative analysis. f At 4 week postoperatively, H&E and Masson staining were performed to evaluate the osteogenic activity in implants. Dotted straight black line indicates the femur-implant junction (I, implant area). Black short arrows, osteocytes. Yellow arrows, chondrocyte-like cells. Black arrows, osteoblasts. Scale bars, 500 µm. Data are presented as the mean ± SEM. **P < 0.01.
Angiogenesis and osteogenesis within FP-TEBs are dependent on CXCR2 activation of ECs
Angiogenesis is a multifaceted process orchestrated by a series of EC-related events. Thus, the following attention was paid to the effects of FP-TEBs on ECs. Considering the critical roles in dictating migration, the chemokines and their receptors in FP-TEBs and TEBs was surveyed with the help of qPCR assays. The initial observation involved comparing mRNA expression of chemokines between TEBs and FP-TEBs incorporating hBMSCs. The findings indicated significantly higher mRNA expressions of CXCL2, CXCL3, CXCL6, and IL8 (also known as CXCL8) in FP-TEBs compared to TEBs (Fig. 2a). To analyze the contribution of theses chemokines, their effects on EC migration were evaluated. In vitro, these ligands were downregulated in human bone marrow stromal cells (hBMSCs) through shRNA lentiviral transfection (Supplementary Fig. 4). Consequently, EC migration toward FP-TEBs fabricated with shRNA-transfected hBMSCs was diminished to different extent. The most significant decrease was observed after CXCL3 or CXCL8 knockdown (Supplementary Fig. 5). In this, we thought that FP-TEBs could facilitate EC migration via various CXC ligands. CXCL3 and CXCL8 might be core ligands, which was expected to be further explored in vivo. Mice lack an CXCL8 homologous gene and prior results have indicated that CXCL1/2/3 may function as the equivalent of CXCL8 in mice. Thus, CXCL2, 3 or 6 was downregulated in mouse bone marrow stromal cells (mBMSCs) by shRNA lentiviral transfection (Supplementary Fig. 4). In vivo, homing of ECs to FP-TEBs with shRNA-transfected mBMSCs was impeded to different extent (Fig. 2b). The most significant decrease was observed after CXCL3 knockdown. Collectively, we suggested that FP-TEBs might promote EC migration through CXCL3. Next, we tried to incorporate recombinant mouse CXCL3 protein into hydrogel and apply the material to modify DBM scaffolds (Supplementary Fig. 6). Based on the release kinetics provided by a mouse CXCL3 ELISA kit, CXCL3 was slowly released until 9 days (Supplementary Fig. 7). Then, CXCL3-loaded DBM were implanted into the femoral defects in mice. Postoperative experiments indicated that compared with blank DBM, the CXCL3-loaded DBM were superior in promoting EC migration and osteogenesis (Fig. 2c and Supplementary Fig. 8). However, the promotive power was significantly lower than that of FP-TEBs. These findings implied that FP-TEBs were extremely likely to facilitate bone regeneration through the synergistic action of multiple cytokines present therein, making it challenging to replicate the therapeutic outcome by loading a singular ligand.
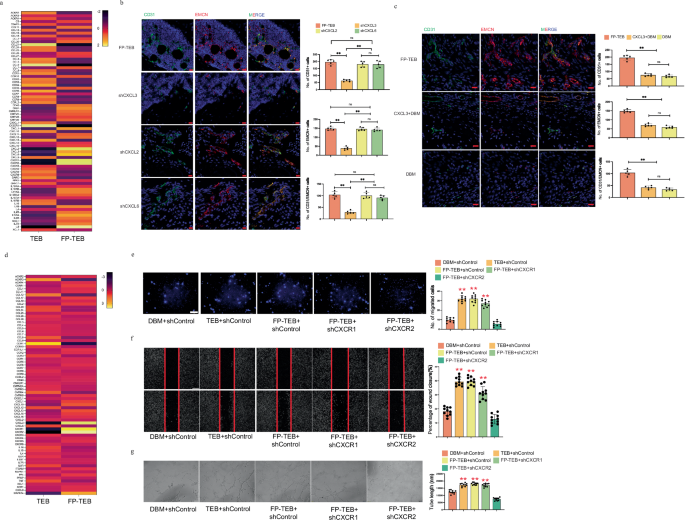
a Quantitative polymerase chain reaction (qPCR) array on chemokine and receptor-related genes of FP-TEBs and TEBs incorporating hBMSCs (n = 3). b Representative immunostaining images with quantification of cell numbers in different FP-TEBs at 2 weeks postoperatively (n = 5). White arrows, staining-positive cells. Scale bar, 10 μm. c Representative immunostaining images with quantification of cell numbers in different implants at 2 weeks postoperatively (n = 5). White arrows, staining-positive cells. Scale bar, 10 μm. d qPCR array on chemokine and receptor-related genes in critical-size bone defects treated with TEBs and FP-TEBs incorporating mBMSCs at 2 weeks postoperatively (n = 3). e Representative images of migrated HUVECs in Transwell systems. Cell quantification is shown as a bar graph (n = 10). Scale bar,100 μm. f Representative images of wound healing assays. The rate of scratch wound closure is shown as a bar graph (n = 10). Scale bar, 200 μm. g Representative images of tube formation assays. The tube length is shown as a bar graph (n = 10). Scale bar, 100 μm. Data are presented as the mean ± SEM. **P < 0.01.
To investigate the difference in receptor expression, TEBs and FP-TEBs incorporating mBMSCs were implanted and subsequent expressions were compared at postoperative day 14 (Fig. 2d). The results showed that the mRNA expressions of CXCL3, CXCR1, CXCR2, MAPK14 were conspicuously higher in FP-TEBs. Together, the significantly elevated expression of both CXCR1 and CXCR2 and their ligands in FP-TEBs highlighted their implication in regulating EC infiltration. In this context, ECs were transfected in vitro with shRNA lentiviral particles to knock down gene expression of cxcr1 or cxcr2 (Supplementary Fig. 9). As revealed by in vitro migration assays (Fig. 2e), FP-TEBs and TEBs significantly augmented EC migration, as compared with DBM. The chemotactic effect of FP-TEBs on ECs was prominently inhibited by knockout of cxcr2, but not cxcr1. Consistent result was obtained from the wound healing (Fig. 2f) as ECs with cxcr2 knockout showed the lowest rate of scratch area closure. Moreover, FP-TEBs showed analogous pro-angiogenic capacity to TEBs, which was abrogated by cxcr2 knockout in ECs, but not cxcr1 (Fig. 2g).
To verify the role of CXCR2 in the vascularization of FP-TEBs, we generated cxcr2tek mice for bone grafting (Fig. 3a). Accordingly, the infiltration of total CD31+ ECs and Type-H ECs induced by FP-TEBs was markedly impaired in cxcr2tek mice, as compared with cxcr2flox mice (Fig. 3b). Concurrently, a reduced presence of newly formed vessels was observed both within and surrounding FP-TEBs implanted into cxcr2tek mice at day 14 (Fig. 3c). Subsequent histological examination indicated a decline in osteogenic activity within these FP-TEBs (Fig. 3d). Quantitative micro-CT analysis demonstrated that BV and Tb.Th were significantly reduced in FP-TEBs in cxcr2tek mice, whereas Tb.Sp exhibited an inverse trend (Fig. 3e). Taken together, these findings suggested that the activation of CXCR2 primarily contributed to the angiogenesis and osteogenesis observed in FP-TEBs.
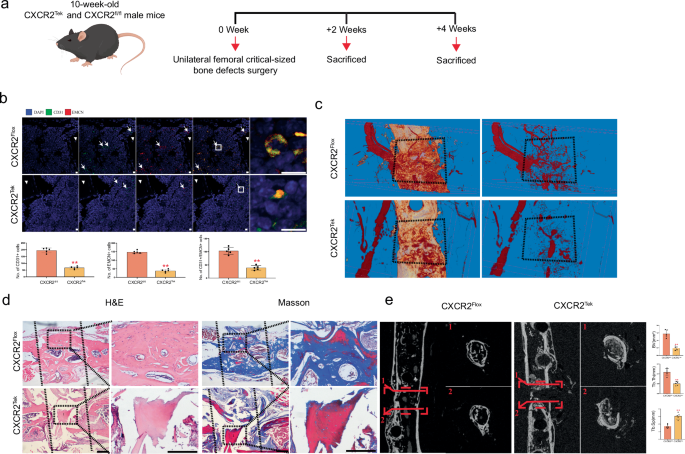
a Schematic diagram of the experiments for assessing the angiogenic and osteogenic effects. b Representative immunostaining images with quantification of cell numbers in critical-size bone defects at 2 weeks postoperatively (n = 5). White arrows, staining-positive cells. Scale bar, 10 μm. c Representative Micro-fill perfusion images of bone defect areas from cxcr2flox and cxcr2tek treated with FP-TEBs at 2 weeks postoperatively. d H&E and Masson staining at 4 week postoperatively. Dotted straight black line indicates the femur-implant junction (I, implant area). Scale bars, 500 μm. e Micro-CT reconstruction and quantitative analysis of the bone volume (BV), Tb. Sp (Trabecular separation), and Tb. Th (Trabecular thickness) of bone defect areas from the cxcr2flox and cxcr2tek mice at 4 weeks postoperatively (n = 5). Dotted straight red line indicates the area of interest for quantitative analysis. Data are presented as the mean ± SEM. **P < 0.01.
Src, Map4k4 and p38 MAPK are indispensable for CXCR2 function
To identify molecules downstream of CXCR2, a set of inhibitors were applied for in vitro migration assays (Supplementary Table 1). Accordingly, pre-treating ECs with LY294002, SP600125 or U0126 only slightly weakened ECs migration towards FP-TEBs. By contrast, the adoption of AZD0530, GNE-495, or SB203580 resulted in a notable decrease in the number of migrated ECs in a manner analogous to cxcr2 shRNA. Then, src, map4k4 or p38 Mapk was knocked down by specific shRNA in ECs and phenocopied cxcr2 shRNA regarding EC migration towards FP-TEBs (Fig. 4a, b, Supplementary Fig. 10).
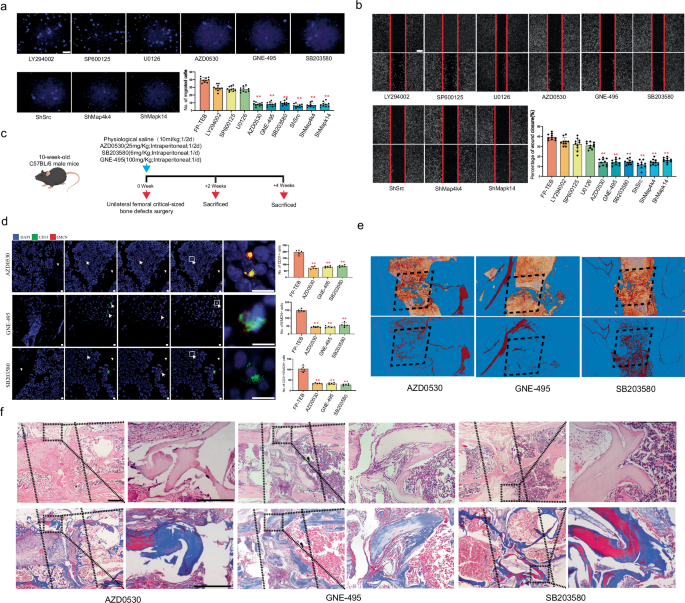
a Representative images of migrated HUVECs in Transwell systems. Quantification of the migrated HUVECs is shown as a bar graph (n = 10). Scale bars,100 μm. b Representative images of wound healing assays. The rate of scratch wound closure is shown as a bar graph (n = 10). Scale bars, 200 μm. c Schematic diagram of the experiments for assessing the angiogenic and osteogenic effects. d Representative immunostaining images with quantification of cell numbers in FP-TEBs implanted into bone defects of cxcr2flox mice, which at 2 weeks postoperatively (n = 5). White arrows, staining-positive cells. Scale bar, 10 μm. e Representative Micro-fill perfusion images of bone defect areas from cxcr2flox mice treated with FP-TEBs at 2 weeks postoperatively. f H&E and Masson staining at 4 week postoperateively. Dotted straight line indicates the femur–implant junction. Scale bars 500 μm. Data are presented as the mean ± SEM. *P < 0.05, **P < 0.01.
To obtain more clear evidence, AZD0530, GNE-495, and SB203580 had been administrated into cxcr2flox mice via intraperitoneal injection after FP-TEBs were implanted (Fig. 4c). All of them echoed the negative effect in cxcr2tek mice, since the infiltration of CD31+ and Type-H ECs was markedly reduced (Fig. 4d). The abrogation of EC infiltration in cxcr2flox mice induced by AZD0530, GNE-495, or SB203580 was found to be associated with impaired vascularization and reduced osteogenic activity in FP-TEBs. Microfil-perfused angiography revealed a diminished vascular network in the central region of the FP-TEBs following injection of AZD0530, GNE-495, or SB203580 (Fig. 4e). Subsequent histological examination indicated a decline in osteogenic activity within these FP-TEBs (Fig. 4f). Quantitative micro-CT analysis demonstrated significantly lower values of BV,Tb.Th and higher Tb.Sp in the FP-TEBs scaffolds after injection with AZD0530, GNE-495, or SB203580 (Supplementary Figure 11).
FP-TEBs promote migration of ECs via CXCR2 and its downstream kinases Src-Map4k4-p38 MAPK
Although we have demonstrated that FP-TEBs regulated EC migration through activation of CXCR2 and its downstream signal molecules, further investigation was required to understand the upstream and downstream relationship among Src, MAP4K4, and p38 MAPK in regulating EC migration. Through in vitro immunofluorescence experiments on ECs, we found that inhibition of MAP4K4 and p38 MAPK did not yield a significant impact on the phosphorylation of Src. However, inhibiting Src significantly reduced the expression of MAP4K4 in ECs. Conversely, inhibiting p38 MAPK showed unconspicuous influence on the expression of MAP4K4. Moreover, the phosphorylation of P38 MAPK was visibly suppressed after blockade of Src or MAP4K4 (Fig. 5a–c). Similar variation trend was further provided by western blot (Fig. 5d). ShRNA targeting Src led to downregultion of MAP4K4, p38 MAPK, but not JNK or ERK1/2. Phosphorylation of p38 MAPK, but not Src, was inhibited by MAP4K4 blockade. Suppresion of p38 MAPK showed no obvious influence on Src and MAP4K4. Together, these findings suggested that CXCR2 was likely to regulate ECs through the Src-MAP4K4-p38 MAPK axis. In vivo, silencing cxcr2 in ECs imposed noteworthy restrictions on the expression of the Src-MAP4K4-p38 MAPK axis, providing corroborative evidence on the up-downstream relationship. Meanwhile, the recruitment of CD31+ ECs and Type-H ECs in FP-TEBs was significantly inhibited by Src, MAP4K4, and p38 MAPK inhibitors, as revealed by immunofluorescence. Src inhibition notably decreased the expression of MAP4K4 and the phosphorylation of p38 MAPK. Blockade of MAP4K4 only exerted remarkable negative effects on the phosphorylation of p38 MAPK, but not Src. However, the inhibition of p38 MAPK did not exhibit any impact on MAP4K4 and Src expression (Fig. 6a–c). Collectively, these findings demonstrated that the infiltration of ECs in FP-TEBs was dependent on CXCR2 and the downstream Src-Map4k4-p38 MAPK axis.
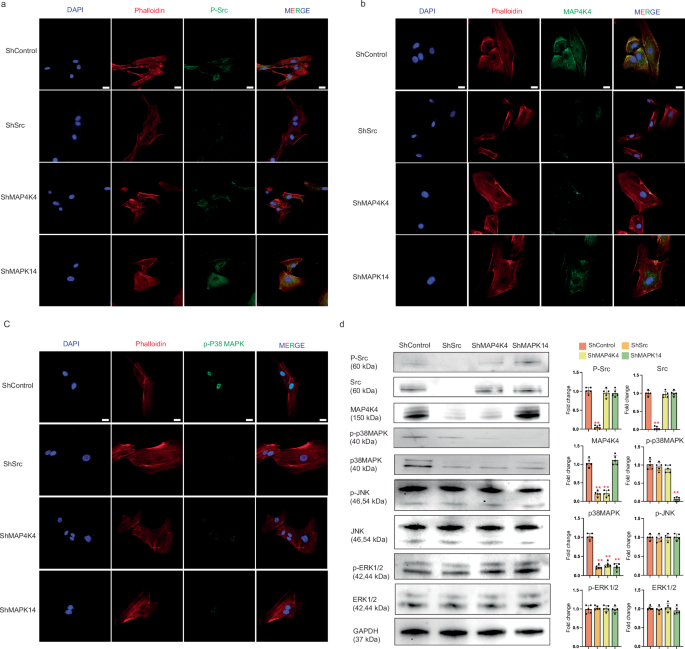
a IHC was performed on HUVECs using antibodies against p-Src and antibodies specific to phalloidin. 488-conjugated Goat Anti-Rabbit IgG secondary antibody was used. The nuclei were stained with DAPI (n = 10). Scale bar, 20 μm. b IHC was performed on HUVECs using antibodies against MAP4K4 and phalloidin. (n = 10). Scale bar, 20 μm. c IHC was performed on HUVECs using antibodies against p-p38 MAPK and phalloidin. (n = 10). Scale bar, 20 μm. d Protein expression in HUVECs transfected with shSrc, shMAP4K4 and shMAPK14 (n = 3). Data are presented as the mean ± SEM. **P < 0.01.
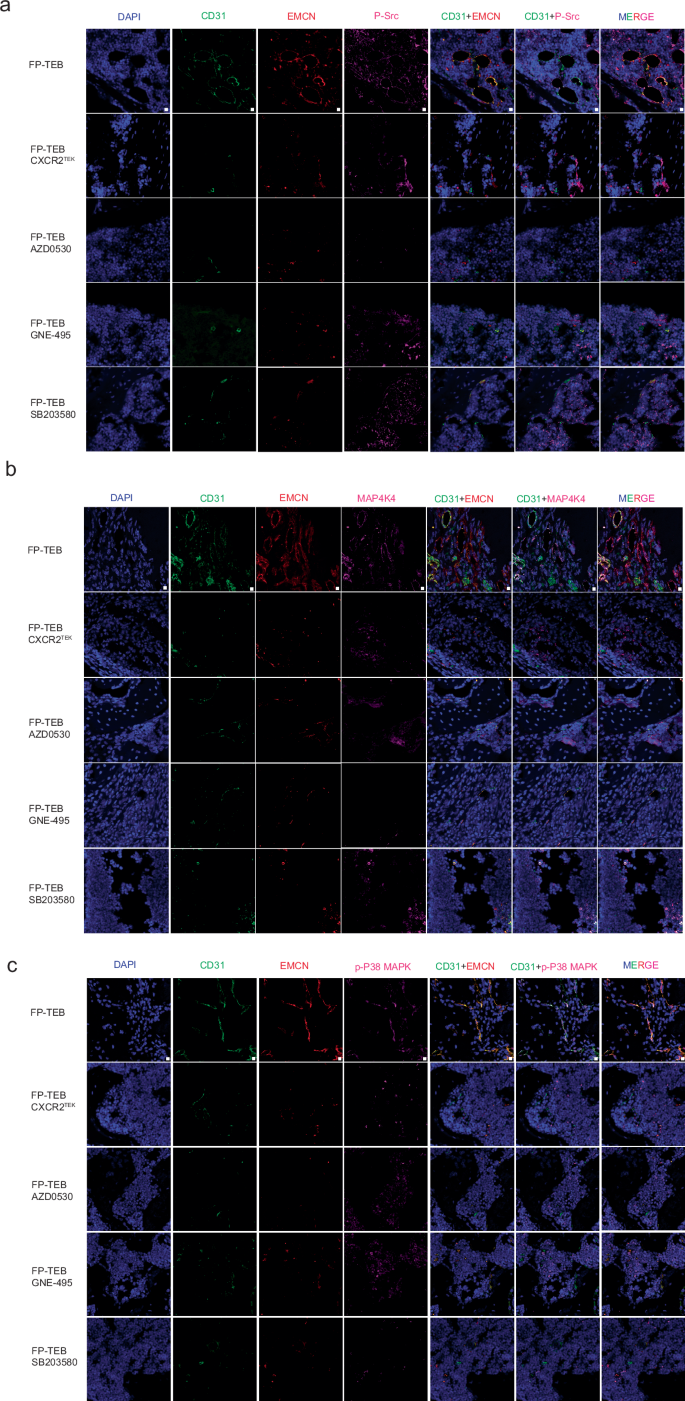
a Representative IHC images of bone defect areas from cxcr2flox mice treated with FP-TEBs at 2 weeks postoperatively. P-Src was labeled with 647-conjugated Goat Anti-Rabbit IgG secondary antibody. CD31 was labeled with 488-conjugated Rabbit Anti-Goat IgG secondary antibody. EMCN was labeled with 555-conjugated Rabbit Anti-rat IgG secondary antibody (n = 10). b Representative IHC images. MAP4K4 was stained with 647-conjugated Goat Anti-Rabbit IgG secondary antibody (n = 10). c Representative IHC images P-p38 MAPK was labeled with 647-conjugated Goat Anti-Rabbit IgG secondary antibody (n = 10). The nuclei were stained with DAPI. Scale bars,10 μm.
Discussion
Accumulating evidence supports the advantages of MSCs-based strategies in repairing LSBD, emphasizing the critical roles of incorporated MSCs2,19. Still, the clinical application of TEBs faces challenges primarily associated with the dependence on viable cells. Other than the aforementioned hurdles, rigorous quality control systems, labs adhering to stringent specifications, and highly skilled technicians are required for construction. TEBs are inapplicable to patients with dysfunctional MSCs, such as the elderly and individuals with autoimmunity diseases or severe underlying conditions like diabetes, osteoporosis, etc. Moreover, recent literature highlights difficulties in retaining MSCs directly delivered to injury sites, as these cells become undetectable within a timeframe too short for self-renewal and differentiation4. Consequently, the therapeutic effects of MSCs in tissue repair are mainly attributed to their paracrine biologics20. Thus, developing composites with effective cellular components rather than viable stem cells holds great promise for strategy innovation21,22.
Targeting this goal, we previously introduced FP-TEBs and their osteogenic potentials were assessed preliminarily in vitro9. In this study, we further reiterated in vivo that FP-TEBs owned impressive osteogenic competence, which was analogous to TEBs and much greater than blank scaffolds. During bone repair, osteogenesis is closely coupled and to some extent, fatefully controlled by the vascularization status17,18. Other than immune-mediated damage and apoptosis, poor hemoperfusion exerts enormous impacts on MSCs following administration. Cells must be within ~200μm of the nearest vessels to acquire sufficient nutrients and oxygen, but it may take too long time for vessel ingrowth, leading to cell vanishment23. However, enhanced osteogenesis in TEBs and FP-TEBs, along with previous evidence of advanced vessel formation in TEBs, strongly suggest that the scaffold loads may ameliorate vascularization. Therefore, we further investigated the angiogenic capacities of FP-TEBs and TEBs, finding a marked increase in vascularization compared to controls.The superiorities of TEBs and FP-TEBs over DBM in angiogenesis, as well as osteogenesis, should be attributed to the loads in scaffolds, more exactly, livable MSCs in TEBs and retained cellular components in FP-TEBs. Intriguingly, the recruitment of Type-H ECs was advanced in FP-TEBs, suggesting that while the angiogenic and osteogenic capabilities of FP-TEBs and TEBs were similar, FP-TEBs exhibited a superior osteogenic process, at least for a specific duration. This disparity may be attributed to the distinct scaffold loads. Living MSCs likely interact with the surrounding microenvironment, altering the secretome of MSCs and promoting the infiltration of Type-H ECs due to therapeutic biologics in the environment24. As with FP-TEBs, a different functional mode had been assumed, since the release kinetics of biologics were controlled to be slow, yielding progressively enhanced effects on host cells.
ECs naturally migrate to injury sites, a process managed by various factors, typically chemokines25. We have previously demonstrated the robust paracrine capability of MSCs, which includes the release of C-X-C motif ligands, amplified by inflammatory microenvironments6. Inspired by this, we examined the levels of chemokines and their receptors in FP-TEBs and TEBs using qPCR assays. Elevated expression of chemokines CXCL2/3 and CXCL6, with the most significant difference observed in IL8, was found in FP-TEBs. In vitro and in vivo experiments revealed that various CXC ligands contributed to FP-TEBs-facilitated EC migration and CXCL3 might be a core ligand. However, the capacity of CXCL3-loaded DBM in promoting EC migration and osteogenesis was significantly weaker than that of FP-TEBs, indicating that FP-TEBs were extremely likely to facilitate bone regeneration through the synergistic action of multiple cytokines present therein. Subsequent qPCR analysis conducted on ECs within the implants in a murine model revealed notable expression levels of chemokine receptors CXCR1 and CXCR2, as well as chemokine MAPK14. These receptors and chemokine have previously been associated with the regulation of EC function, thus suggesting their potential involvement in bone repair facilitated by FP-TEBs.Inhibiting CXCL1/2/3 or their receptor, CXCR2, markedly impaired vessel growth and HUVEC migration induced by carcinoma cells14. Similarly, CXCL5 exhibited chemoattractant effects on HUVECs and regulated angiogenic properties via CXCR226. CXCL8, a potent activator of CXCR1 and CXCR2, had significant impacts on endothelial lineage cells27. Impressively, these chemokines share one high-affinity receptor, CXCR2. Collectively, it was logical to assume that CXCR1 and CXCR2 play vital roles in the vascularization of FP-TEB. In line with this, a previous study suggested that for ECs, knockdown of cxcr1 and cxcr2 by shRNAs resulted in a significant and parallel decrease in cell migration28. Here, we further demonstrated that the migration of ECs, including Type-H ECs, vascularization, and bone repair in FP-TEBs were dependent on CXCR2, but not CXCR1. CXCR1 and CXCR2 are closely related receptors sharing 76% sequence homology and transduce signals through a G-protein-activated second messenger system29,30. They can both recognize CXCL members, such as CXCL6 and CXCL8. Nevertheless, CXC chemokines with high levels in FP-TEBs, including CXCL1/2/3 and CXCL5, mainly function via binding to CXCR2, but not CXCR131,32. Additional evidence interpreting such difference lies in the regulatory modes, which are distinct between CXCR1 and CXCR2 regarding receptor phosphorylation, internalization, and transactivation33.
CXCR2 is highly expressed by neutrophils, constitutively expressed by ECs and acts as an adaptor in numerous signaling pathways that positively regulate downstream kinase cascades associated with EC migration and angiogenesis34,35. Hu et al. found that Kaposi’s sarcoma-associated herpesvirus-encoded miR-K12-3 promoted migration and invasion of ECs through a CXCR2-Akt signaling axis36. Miyake et al. demonstrated that CXCL1 stimulated ECs, sustained cellular viability and accelerated angiogenesis via CXCR2 and downstream extracellular regulated protein kinase (Erk)1/237. At the gene level, cxcr2 activation in endothelial lineage increased the expression of Src and phosphorylated c-Jun N-terminal kinases (JNK) and Erk1/237,38. Regarding cell migration, other potential molecules downstream of CXCR2 included p38 MAPK and MEK1/239. Given these findings, CXCR2-mediated EC migration might engage one or more signaling pathways, such as PI3K/Akt, JNK-eNOS, MEK/Erk, p38 MAPK, MAPK, and FAK/Src. Therefore, we used pathway inhibitors in in vitro migration assays and found that Src, MAP4K4, and p38 MAPK were the primary effectors downstream of CXCR2 in EC migration toward FP-TEBs. Src, a non-receptor tyrosine kinase, plays a critical role in cell-cell junction, cytoskeleton contraction, and vascular hyperpermeability, necessary for cell polarization during directed migration40,41. Blocking Src reiterated the outcome of cxcr2 knockout, nearly abrogating homing of Type-H ECs, vascularization, and bone repair. The decrease in p-Src expression observed within the region of FP-TEBs in cxcr2tek mice further supported the involvement of CXCR2 in EC migration through Src.
The classical three-tiered MAPK cascades involve MAP kinase kinase kinase (MAP3K) activating MAP kinase kinase (MAP2K), which, in turn, activates MAPK, leading to downstream cellular responses. MAP3K is often activated by small guanosine-50-triphosphate (GTP)-binding protein, but in some pathways, it can be activated by another kinase known as a MAP kinase kinase kinase kinase (MAP4K)42,43. MAP4K4, a serine/threonine kinase belonging to the MAP kinase family, plays a crucial role in HUVEC migration and inflammation44,45. The importance of MAP4K4 in normal embryonic development is apparent as its absence in the entire organism or specifically in endothelial cells leads to non-survival45. The potential functional mechanisms of MAP4K4 include the activation of JNK, p38 MAPK, and ERK1/211,45. Numerous studies have extensively shown that MAP4K4 modulates cell proliferation and apoptosis via the JNK pathway46. The role of MAP4K4 in ERK signaling appears cell type-specific. For example, MAP4K4 phosphorylates ERK in skeletal muscle cells isolated from human biopsies, but not in rodent C2C12 myoblast47. Our study showed that downregulation of Src expression resulted in a notable decrease in MAP4K4 and p38 MAPK expression, as demonstrated in in vivo and in vitro experiments. Conversely, inhibition of MAP4K4 solely affected p38 MAPK expression. The p38 MAPK cascades are paramount in regulating diverse endothelial functions responsive to external and internal stimuli, including growth factors, stress, and cytokines48,49. Activation of p38 MAPK in ECs leads to actin remodeling, angiogenesis, and DNA damage response, significantly impacting vascular homeostasis47,49. Our findings provided compelling evidence for the indispensability of p38 MAPK in CXCR2-mediated EC infiltration into FP-TEBs, as well as the development of newly formed vasculature and bones. In conclusion, during the treatment of LSBD, FP-TEBs promoted EC infiltration, vascularization and bone repair via CXCR2 and its downstream kinases Src-MAP4K4-p38 MAPK.
Nonetheless, some limitations exist in this study. Firstly, due to experimental constraints, only cxcr2 was conditionally knocked out in ECs. Identification of src, map4k4 and p38 was achieved via injection of selective inhibitors, introducing some confidence limitations. Secondly, the proportion of Type-H ECs in total ECs was low, making sorting of Type-H ECs challenging, especially in cxcr2tek mice. Since ECs were fundamental components of vessels, biological assessments mainly focused on sorted ECs. Finally, unlike Src for CXCR2, where the relationship seems to be unique, signal molecules associated with CXCR2 remain more complicated regarding EC migration towards FP-TEBs. Further experiments based on high-throughput proteomics and genomics are needed to gain deeper insights.
In summary, we provide primary evidence that the novel FP-TEBs demonstrate comparable capabilities to TEBs in vessel and bone reconstruction. The angiogenesis and osteogenesis within FP-TEBs require the activation of CXCR2 and its downstream kinases Src-MAP4K4-p38 MAPK. These findings advance the development of cell-free tissue engineering approaches and provide light into formulating homing-based strategies for tissue regeneration.
Methods
Cell isolation and expansion
All protocols involving human subjects were approved by the Ethics Committee, Southwest Hospital, Third Military Medical University, with all subjects providing informed consent. This study was conducted in strict accordance with the principles outlined in the Declaration of Helsinki for research involving human subjects. Human bone marrow MSCs (hBMSCs) were purchased from Cyagen Biosciences (Cat No: HUXMA-01001). Mouse mesenchymal stem cells (mBMSCs) were isolated and cultured as previously described50. Briefly, bone marrows were extracted from femurs by resecting the epiphyses and flushing the shaft with cold phosphate buffered saline. Cells were collected by centrifugation and resuspended in the culture media. After 24 h, non-adherent cellswere discarded and the culture media were changed every 48–72 h. When reaching confluence of more than 80%, cells were trypsinized and passaged for 3 times before use. hBMSCs and mBMSCs were cultured in basic culture medium containing Dulbecco’s modified Eagle’s medium/F12 (DMEM/F12; 1:1; Hyclone) supplemented with 10% fetal bovine serum (FBS; Gibco) and 100 U/ml penicillin/streptomycin (Gibco). Human umbilical vein endothelial cells (HUVECs) were purchased from Cyagen Biosciences (Cat No: HUVEC-20001). HUVECs were cultured in endothelial cell medium (ScienCell Research Laboratories) supplemented with 10% FBS and 100 U/ml penicillin/streptomycin. The media were changed every other day. When reaching 80–90% confluence, cells were digested using 0.25% trypsin-EDTA (Gibco) and passaged. HUVECs at passage 3, hBMSCs and mBMSCs at passage 4 were harvested for use.
Gene interference
HUVECs, mBMSCs, and mBMSCs were infected with lentivirus particles encoding the corresponding short hairpin RNAs (shRNA; Santa Cruz Biotechnology) according to the manufacturer’s instructions. Clones expressing the virus were selected by their resistance to puromycin (Sigma-Aldrich). The interference efficiency was confirmed by western blot (Supplementary Figs. 4, 9, and 10).
Preparation of implants and animal modeling
For in vitro studies, allogenic DBM scaffolds (Datsing Bio-Tech Co., Ltd.) were cut into blocks (1 × 1 × 0.5 cm; 0.417 g/block) and immersed in DMEM/F12 medium overnight at 4 °C. Human TEBs were fabricated by dropwise instillation of an aliquot (20 µl) of single-cell suspension of hBMSCs (1 × 107/ml) onto two opposite surfaces of the scaffold. After 2 h of incubation at room temperature, media were added and replaced every 2 days for 10 days. For in vivo use, DBM were prepared by coring plugs from the subchondral regions of young Yunnan miniature pigs and washing with water and detergents to remove adipose tissue, calcium and protein, as previously described50. After sterilization by irradiation, DBM (0.2 × 0.2 × 0.2 cm) were immersed in DMEM/F12 overnight for later use. Mouse TEBs were fabricated by seeding mBMSCs onto DBM (1 × 105 cells/scaffold) and culturing for 10 days.
Recombinant mouse CXCL3 protein (HY-P7153, MedChemExpress) was combined with a hydrogel (Pluronic® F-127, Sigma) and applied to modify DBM to fabricate CXCL3-loaded DBM. Briefly, 25%(w/v) Pluronic F-127 powder was slowly dissolved in the precooled phosphate-buffered saline (PBS) buffer solution by magnetic stirring at 4 °C overnight, then filtered with a 0.22 μm filter (SLGPR33RB, Merck Millipore), and maintained at 4 °C for use. Mouse CXCL3 proteins were encapsulated within PF-127 solution with 100 ng, and the mixture was blended with DBM and stored at 4 °C. In vitro release profile of CXCL3 from the scaffold was evaluated over time. The CXCL3-loaded scaffolds were placed in test tubes and immersed in 2 mL PBS at 37 °C under a constant shaking of 30 rpm. At each time point, the supernatant was collected and replaced with an equal amount of fresh PBS. The amounts of released CXCL3 were quantitatively analyzed using a mouse CXCL3 ELISA kit (MCX130, R&D) to plot the release profile.
FP-TEBs were prepared by freeze-drying human or mouse TEBs. Briefly, TEBs were washed with sterile PBS to remove culture medium and cellular debris. Afterthat, TEBs were transferred into a 96-well plate and precooled in a −80 °C freezer for 2 h. Meanwhile, the lyophilizer was turned on for precooling. The frozen materials were then placed into the lyophilizer for freeze-drying over a period of 48 h under sterile conditions (−80 °C, 3 × 10−2 m bar) and then stored at 80 °C for 3 months. Using a scanning electron microscopy (Phenom Pro Desktop SEM, thermo fisher), the viability of mBMSCs was corroborated on TEBs and the pores of FP-TEBs were covered by cell debris and deposited ECM (Supplementary Fig. 12).
Tek-cre; cxcr2floxp/floxp mice (hereafter cxcr2tek mice) and cxcr2floxp/floxp (hereafter cxcr2flox mice)mice mice (C57, 10 week-old, weighing 18.0–20.5 g) were purchased from Cyagen Biosciences. Efficient cxcr2 knockout was confirmed by Western blot of freshly isolated ECs (Supplementary Fig. 13). C57 male mice (10 week-old, weighing 22.5–23.5 g) were designated as recipients for bone grafting. Femoral critical-sized bone defects (2 mm in length) were created according to the procedure previously reported and implanted with FP-TEBs, TEBs or DBM51,52. Briefly, mice were anesthetized by 2–3% isoflurane and orally intubated with a 22 G i.v. catheter, and artificially ventilated with a respirator, the lower limb was shaved and disinfected. A small incision was made on the superior lateral aspect of the distal femoral condyle. Blunt dissection was implemented within the intermuscular space to expose the femoral shaft and the periosteum was carefully stripped from the trochanter to the condyle, preserving the articular capsule distally. Then, a segmental defect was created using a steel plate. On the lateral surface of the femur, a marker was firstly made at 2 mm distal to the lower margin of tuberosity. Using the maker as a navigation, the plate was accurately located and four holes were drilled through the femur along the plate holes. The plate was secured to the femur with four steel screws. 2 mm of the middle femoral shaft was removed using a dental grinding drill. After washing, the scaffold was implanted into the defect site and fixed with decussate sutures. Muscle and skin were closed layer by layer. Postoperatively, all mice were awakened, and buprenorphine hydrochloride was injected subcutaneously every 12 h until 24 h (1 mg/Kg) for analgesia. General health and activity were monitored daily. Cxcr2flox mice receiving implantation of FP-TEBs were randomly grouped according to the diverse postoperative treatments (Supplementary Table 2). The mice were euthanized with 60% carbon dioxide before sampling. All animal experiments were approved by the Animal Care and Use Committee of the Third Military Medical University.
Immunofluorescent staining
For cell immunofluorescence staining in vitro, HUVECs were first washed with ice-cold PBS. Subsequently, they were fixed in a 4% paraformaldehyde solution for 15 min and incubated in a 0.1% Triton solution at room temperature for 15 min. After three additional washes with PBS, cells were blocked using a 5% fetal bovine serum solution at 4 °C for 30 min. Primary antibodies targeting p-Src (1:200, Abclonal), p-P38 MAPK (1:200, Abclonal), MAP4K4 (1:100, Cell Signaling Technology), and Rhodamine Phalloidin (1:400, Abclonal) were incubated with cells overnight at 4 °C. Then, cells were stained with secondary antibodies (488-conjugated Goat Anti-Rabbit IgG,1:200, Abclonal) for 1 h and DAPI for 10 min. For each sample, five separate sections were randomly selected from at least 10 sections and subjected to a confocal laser scan microscope (Leica Biosystems).
For tissue immunofluorescence staining, five mice from each group were euthanized with 60% carbon dioxide before sampling, implants were collected at 14 days postoperatively. After fixation in 4% paraformaldehyde and decalcification with EDTA, frozen sections (7 mm thick) were prepared, permeabilized with 0.3% Triton X-100 for 10 min, and blocked with normal donkey serum (1:50; Huayueyang Biotechnology) at room temperature. Slides were incubated with primary antibodies rabbit anti-p-Src, p-P38 Mapk (1:100, Abclonal), MAP4K4 (1:100, Cell Signaling Technology), goat anti-CD31 (1:50, R&D Systems), rat anti-Emcn (1:100, Santa Cruz) overnight at 4 °C. Then, the sections were stained with secondary antibodies 647-conjugated Rabbit Anti-Rabbit IgG (1:200, Abclonal), 555-conjugated Rat Anti-Rat IgG (1:200, Abclonal), and 488-conjugated Mouse Anti-Goat IgG (1:200, CST) for 1 h and DAPI for 10 minuts. For each sample, five separate sections were randomly selected from at least 10 sections and visualized using a confocal laser scan microscope (Leica Biosystems).
Micro-CT reconstruction
To evaluate vascularization status within and around implants, three mice in each group were randomly chosen for Microfil perfusion at postoperative day 14. Briefly, mice anesthetized by 2–3% isoflurane and positioned supine to allow access to the rib cage. After skin preparation with chlorhexidine, a midline incision across the thorax and abdomen was made, the heart and adjacent great vessels were exposed. Then the thoracic aorta was punctured and the inferior vena cava was incised, followed by sodium heparin injection until the liver turned pale. The lower extremities were perfused with Microfil working solution. The whole femurs were excised and fixed with 4% polymethylaldehyde for 24 h and then scanned by Micro-CT (Skyscan 1272; Bruker microCT). To evaluate the osteogenic capacity of the implants, five mice in each group were randomly chosen for Micro-CT scanning at postoperative week 4. Briefly, mice were euthanized, and the whole femurs were excised and fixed with 4% polymethylaldehyde for 24 h and then scanned by Micro-CT (Skyscan 1272; Bruker microCT). The raw data were processed with NRecon 1.7.1.0, CT analyzer 1.16.4.1 for noise removal, and CTvox 3.3 for 3D reconstruction and rendering.
Histological observation
Five mice in each group were euthanized with 60% carbon dioxide at 4 weeks postoperatively. The grafts were excised, decalcified with 10% EDTA, dehydrated in graded alcohol solutions and embedded in paraffin. Samples were sectioned (6-mm in thickness) and subjected to hematoxylin and eosin (H&E) and Masson’s trichrome staining as described before50. Tartrate-resistant acid phosphatase (TRAP) staining was performed to evaluate the osteoclast activity and bone resorption. Sections were incubated with TRAP stain solution (Sigma-Aldrich) according to the manufacturer’s instructions. Photomicrographs were obtained using a light microscope (Olympus).
RT-PCR and qPCR assays
Total RNA of implants harvested from bone defects were isolated following the manufacturer’s protocol (TriPure, Roche). SYBR green quantitative PCR (iQ SYBR green supermix, Bio-Rad) was performed on the Bio-Rad CFX97. Primer sequences are listed in Supplementary Table 3. The qPCR mixture (10 μl) is composed of 5 μl Roche FastStart Universal SYBRGreen Master (2 ×), 0.75 μl each primer (10 μM), 3 μl ddH2O and 0.5 μl template. Initial enzyme activation was performed at 95 °C for 10 min, followed by 40 cycles of denaturation at 95 °C for 30 s and annealing at 60 °C for 30 s. The melting curve was generated ranging from 60 °C to 95 °C to determine the specificity of amplification. To investigate the expression difference of chemokines between FP-TEBs and TEBs, human Chemokines and receptors qPCR assays (Wcgene Biotech) were performed on human FP-TEBs and TEBs according to the manufacturer’s protocols. To compare the cell receptors between FP-TEBs and TEBs, implants harvested at 14 days were subjected t o mouse Chemokines and receptors qPCR assays (Wcgene Biotech).
Migration assays in vitro
To prepare conditioned media, human TEBs, FP-TEBs and DBM (Datsing Bio-Tech Co., Ltd.) were cut into small blocks (0.5mm3) and added into endothelial cell medium (ScienCell Research Laboratories) supplemented with 10% FBS and 100 U/ml penicillin/streptomycin. Migration assays were performed in Transwell inserts (8-mmpores; Corning). To exclude the influence of cell proliferation on migration, HUVECs were treated with 10 μg·mL−1 cytochalasin C for 2 h. Then, cells were pre-incubated with inhibitors (Supplementary Table 1) and loaded into the upper chamber (5 × 104 cells/test). Conditioned media were loaded into the lower chamber. ECs were allowed to migrate at 37 °C. After 12 h, cells on the upper side of the filter (non-migrating cells) were removed with a cotton wool swab (NorgenBiotek). Migrated cells on the lower face were washed with PBS and fixed with 4% paraformaldehyde (Boster Biologic Technology). The membrane was then placed onto glass slides with the bottom side upward. Migrated cells were stained with DAPI and subjected to microscopy. For each group, the number of migrated cells was counted on 10 random high-power fields (×200 magnification) and averaged.
For the wound healing assay, HUVECs were seeded and cultured in 6-well plates (1 × 105/well) to reach the confluent monolayer. To exclude the influence of cell proliferation on migration, 10 μg·mL−1 cytochalasin C were added for 2 h. Cells were pre-treateed with inhibitors (Supplementary Table 1). Then, cells were scraped using a 200 μL pipette tip and washed with PBS to clear cell debris and suspension. Afterthat, different types of media were added, including serum-free medium and conditional media. Microscopic images were captured at the same position of the wound area at 0 and 12 h. Migration ability was measured by the rates of scratch wound closure using the ImageJ software (National Institutes of Health).
Tube formation assay
HUVECs were starved overnight using culture medium supplemented with 0.2% serum. Then, the 96-well plates were coated with 50 µl of matrigel (Corning) and incubated at 37 °C for 30 min to promote jelling. HUVECs were resuspended in 50 µl EGM-2 medium and added to each well (103 cells/well) with conditioned media (50 µl/well). After incubation at 37 °C for 12 h, images from at least ten random areas were captured for each group. The tube branch length was measured by software ImageJ (National Institutes of Health, USA) with the ‘Angiogenesis Analyzer’ plugin. Tube formation assay was repeated at least three times.
Western blot
For in vitro experiments, cells were lysed with SDS lysis buffer (100 mMTris at pH8.0,10% glycerol, and 1%SDS), and protein concentration was determined using a NanoVue spectrophotometer (GE Healthcare). For in vivo experiments, protein was extracted from implants harvested from bone defects using radioimmunoprecipitation assay lysis buffer (Beyotime Biotechnology), and protein concentration was determined using a NanoVue spectrophotometer (GE Healthcare). A total of 30 mg of protein lysates of each sample was separated by SDS-PAGE (80 V, 120 min; Beyotime,) and transferred to PVDF membranes (250 mA3 60 min; Millipore). After blocking with 5% milk, membranes were incubated overnight at 4 °C with primary antibodies, including anti-CXCL2, anti-MAP4K4, anti-Runx2 (1:1000; Cell Signaling Technology), and anti-CXCR1, anti-CXCR2, anti-CXCL6, anti-CXCL8, anti-ALP, anti-OCN, anti-SRC, anti-pSRC, anti-pP38 MAPK, anti-P38 MAPK, anti-JNK, anti-pJNK, anti-ERK, anti-pERK (1:1000; Abclonal), and anti-Alp (Invitrogen,1:1000) followed by incubation with goat anti rabbit secondary antibody (1:10,000; Abclonal) at room temperature for 1 h. Signals were detected by ECL (Kirkegaard & Perry Lab). GAPDH (1:10,000, Abclonal) and beta Actin (1:10,000, Abcam) was used as the loading control. Western blot was repeated 3 times for each sample. Uncropped Western blotting images were provided in Supplementary Fig. 14, where the size markers were labeled. All blots and gels originated from a single experiment and were treated concurrently.
Ethics approval and consent to participate
All protocols were approved by the Ethics Committee Southwest Hospital, Third Military Medical University, with all subjects providing informed consent (AMUWEC20210617).
Statistical analysis
Statistical analyses were performed using GraphPad Prism 10 software. The results were presented as the mean ± standard error of the mean (SEM). For comparisons between two groups, unpaired two-tailed Student’s t tests were conducted. For comparisons among multiple groups, one-way or two-way analysis of variance (ANOVA) with the Bonferroni post hoc test was performed. P < 0.05 were considered significant.

Responses