Integrative systems biology framework discovers common gene regulatory signatures in mechanistically distinct inflammatory skin diseases

Introduction
Nearly 25% of the world’s population is affected by non-communicable chronic inflammatory diseases1. Some of the inflammatory skin diseases include acne, atopic dermatitis (AD), actinic keratoses (AK), psoriasis (PS), hidradenitis suppurativa (HS), and three types of rosacea (RS) share specific immune regulator activities with psoriasis1,2,3. Most of these diseases manifest autoimmune signatures on a temporal basis. Psoriasis is one of the best-described inflammatory skin diseases that affects approximately 1–3% of global adults4. Psoriasis patients manifest comorbidities such as diabetes, metabolic disorders, and severe cardiovascular disease5. Epidermal keratinocytes are the major targets in various skin diseases including HS & PS6,7,8,9. These keratinocytes act as the disease driver in corroboration with immune cells, specifically T helper 17 cells (Th17), which are also considered important in the pathogenesis and treatment of these and various other disease10,11. Importantly, targeting Th17 cell signaling and several interleukin cytokines including IL-23, IL-17A, and IL-17F have improved long-term remissions by 85–100% in psoriasis patients5,11. Similarly, AD is an immune-mediated disease that is prevalent in approximately 25% of children and 10% of adults12. Likewise, HS has a distinct anatomical etiology including the occurrence of inflammatory nodules, abscesses, and pus-draining sinus tracts at the places where skin rubs against each other like the underarm and groin10. The treatment regimens for AD and HS have limited effectiveness and cannot serve effectively the majority of moderate to severe disease patients. Therefore, understanding the shared and unique genetic signatures, activated pathways, and regulators through innovative systems biology and integrative multi-omics can be highly significant and rewarding.
The widespread availability of bulk transcriptomics datasets presents an invaluable opportunity for systems biologists to integrate transcriptomics with other omics layers, including biological networks. This approach enables a comprehensive and extrapolated investigation of diseases, particularly immune-mediated disorders, where the underlying molecular pathways remain incompletely understood13. Network sciences have been applied to diverse biological domains to identify the associated biomolecules in disease, cancer, infections, and other stress conditions. Briefly, biological networks are graphical representations containing nodes (genes/proteins) and edges (links) for a specific condition14,15. These networks tend to change their interacting partners in different scenarios including normal/healthy and disease response16. Network-based systems biology analytics has proven a great tool to unravel the structural properties of biological networks and highlight the significant contributors to disease pathogenesis and organismal development across different systems13,17,18,19,20,21,22. In this study, we designed an integrative systems biology framework to identify the significant regulators across several inflammatory skin diseases. We exploited the gene co-expression network (GCN)- and protein–protein interaction (PPI)-based networks to identify shared genes and protein components in eight skin diseases with relevant functional implications. Further, we identified 55 high-priority proteins (HPPs) with increased network indices, which are also associated with immune-mediated pathways. Finally, we explored the candidate drug–gene interactions to highlight some therapeutic-relevant target selections for developing putative treatment strategies. In summary, our integrative framework unravels the data-based regulatory signatures and activated molecular pathways across inflammatory skin diseases.
Results
A network science-based framework to prioritize genes/pathways from transcriptome datasets
To study the shared and unique genetic signatures, regulators, and prioritizing genes in different chronic inflammatory skin diseases, we implemented an integrative multi-omics framework utilizing the transcriptome datasets and extrapolating the network-based analysis pipeline. In this framework, 11 different steps are followed to identify the most appropriate drug–gene interactions (Fig. 1). 1. First, we extracted publicly available transcriptome datasets in eight different inflammatory skin diseases (namely: acne, atopic dermatitis (AD), actinic keratoses (AK), psoriasis (PS), hidradenitis suppurativa (HS), and three types of rosacea (RS)) as described in the methods. 2. Subsequently, we performed the gene co-expression network construction and analysis for all eight disease datasets through WGCNA23. We achieved eight disease-specific networks ranging from 320 nodes with 264 edges in Acne to 5465 nodes with 132,849 edges in PS. Which demonstrated the challenges in the handling of multivariate transcriptomes. 3. The third step was to perform DEG analysis on all eight disease datasets with the same threshold parameters24. We observed some similarities and differences in expression profiles among eight diseases. 4. To establish the disease-specific protein–protein interaction network, we next combined the results from co-expression networks, DEGs, and the largest publicly available human protein–protein interaction network from the STRING database25. Finally, we had a collection of eight disease-specific protein–protein interaction networks ranging from 90 nodes with 90 edges in Acne to 3622 nodes with 33,173 edges in PS. 5. Based on the network explosibility, a comparable number of nodes and interactions, as well as power-law distribution of eight networks, we selected four disease-specific interaction networks including AD, PS, HS, and RS for further analysis. 6. Afterwards, we performed network centrality analyses on all four filtered networks to identify the most important players in each disease13. Specifically, we analyzed the networks by betweenness centrality (bottlenecks), degree (hubs), eigenvalue (combined effect of degree and betweenness), information centrality (ability to pass stimuli information), and inner core proteins identification by the weighted k-sell decomposition method. 7. With that, we obtained a collection of high-priority proteins (HPPs) in these four inflammatory skin diseases. When comparing the appearance of these HPPs in all four diseases, we identified shared and unique HPPs in each disease. 8. Then, we mapped the HPPs in significantly activated/inhibited signaling pathways in these four skin diseases. Interestingly, we found some highly activated signaling pathways are shared among all four diseases. Additionally, we identified the most important regulators shared among four diseases and mapped them against their regulated pathways. 9. Moving forward, we identified the HHPs interacting with drugs from the publicly available databases DGIdb26. These drug–gene (HPP) interactions can provide significant information about the potential therapeutic options for a disease or class of diseases. 10. From these data, we propose that these drugs can be repurposed for treating several inflammatory skin diseases either alone or in combination. 11. All the predictive outcomes including the role of genes, pathways, HPPs, drug–HPP interactions, and other predictions are required to be validated experimentally for each disease condition. Taken together, we proposed a modular framework that can be applied to any polygenic chronic inflammatory disease to refine the regulator identification, pathway association, and drug–gene prediction for the alternative therapeutic targets in any immunogenetic-related disease.

A Schematic representation of the implemented integrative multi-omics framework utilizing the transcriptome datasets and extrapolating the network-based analysis pipeline. Eleven different steps need to be followed to identify the most appropriate drug–gene interaction for any immunogenetic-related disease.
Different chronic inflammatory skin diseases display unique and conserved transcriptome activity
The conventional transcriptome-based analysis demonstrates the expression behavior in disease for the corresponding cohort of samples. To begin, we identified the total number of DEGs from the transcriptomics (RNA-Seq and microarray) datasets in inflammatory skin diseases including acne, AD, AK, CD, HS, ICD, PS, and three types of RS; erythematotelangiectatic rosacea (ER), phymatous rosacea (PhyR), and papulopustular rosacea (PapR) with a threshold (log2FC ≥ |1|; FDR < 0.05). Based on the transcriptomics platform and diseases, we identified a diverse number of DEGs per disease (Fig. 2A and Data S1). For example, ICD has 65 up- and 47 down-regulated genes, whereas most DEGs were discovered in PS disease with 1763 up- and 3243 down-regulated genes (Fig. S1), respectively. Whereas, acne, CD, and HS have a similar breakdown of DEGs (Data S1). Afterward, we performed the functional analysis for DEGs in each disease to explore the activated/inhibited pathways in those datasets. We report that the Th1 pathway, dendritic cell maturation, Th17 activation pathway, interferon signaling, Th2 pathway, IL-8 signaling, GP6 signaling pathway, CD28 signaling in T helper cells, HOTAIR regulatory pathway, NF-κB signaling are some of the significant activated canonical pathways in these chronic inflammatory skin diseases (BH P-value < 0.05; Fig. 2B and Data S1). Interestingly, we found that the senescence pathway, 1L-7 signaling, and IL-13 signaling are activated only in acne and AK, whereas B Cell receptor signaling was activated in HS, PapR, and CD.
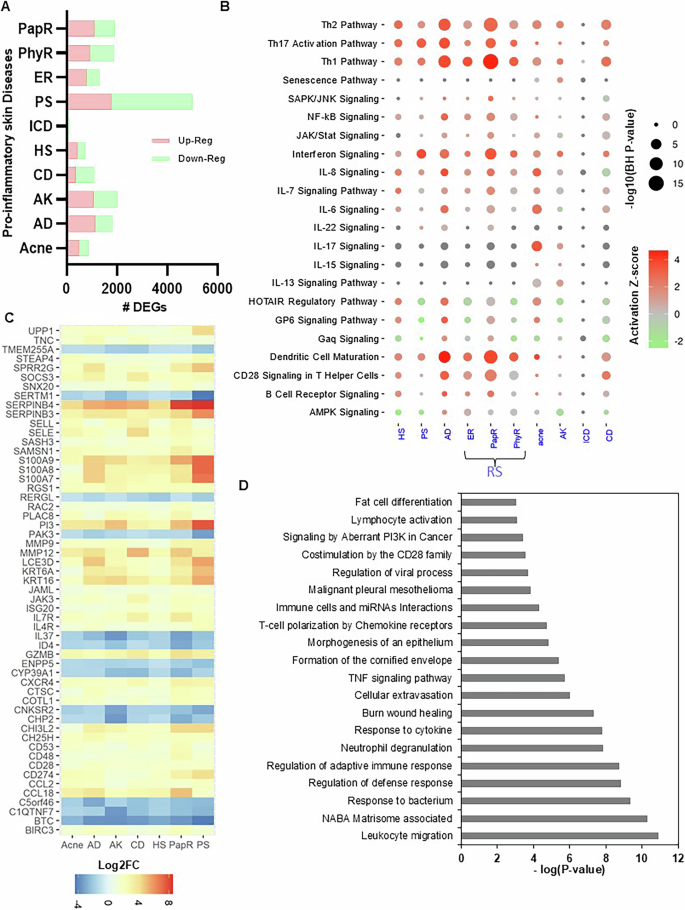
A The total number of differentially expressed genes (DEGs) in inflammatory skin diseases including acne, atopic dermatitis (AD), actinic keratoses (AK), contact dermatitis (CD), hidradenitis suppurativa (HS), irritant contact dermatitis (ICD), psoriasis (PS), and three types of rosacea (RS; erythematotelangiectatic rosacea (ER), phymatous rosacea (PhyR), and papulopustular rosacea (PapR) with a threshold (log2FC ≥ |1|; FDR < 0.05). Upregulated DEGs are marked in light red and down-regulated DEGs are marked in light green. B Significantly activated and inhibited canonical pathways in different inflammatory skin diseases (BH P-value < 0.05). C Differential expression profile of total common DEGs (55) in eight inflammatory skin diseases. D The enriched functional pathways and ontologies of 55 common DEGs across eight inflammatory skin diseases (P-value < 0.05).
To identify the common transcriptome activity among each disease, we performed a commonality analysis. We found that 55 DEGs (including CD28, CD48, CD53, ID4, IL37, IL4R, IL7R, JAK3, KRT16, KRT6A, PI3, S100A7, S100A8, S100A9, SERPINB3, SERPINB4, and UPP1) are common in seven inflammatory skin diseases, except irritant contact dermatitis (ignored due to significantly a smaller number of DEGs reported) (Fig. 2C and Data S1). These are the core transcriptome signatures across diseases and are involved in leukocyte migration, NABA Matrisome associated, response to the bacterium, regulation of defense response, regulation of adaptive immune response, neutrophil degranulation, cytokine response, burn, wound healing, cellular extravasation, TNF signaling, formation of the cornified envelope, morphogenesis of epithelium, T-cell polarization by chemokine receptors, Immune cells, and miRNAs interactions, costimulation by the CD28 family, signaling by aberrant PI3K in cancer, and lymphocyte activation (P-value < 0.05; Fig. 2D and Data S1).
These initial results suggest that there is a huge complexity among chronic inflammatory skin diseases with only a few shared and distinct genes and associated pathways. Thus, we explored the system biology-based methods to unravel intricate relationships, novel gene regulators, and master regulators of pathways altered in these diseases.
The correlation method predicted novel genes involved in four (AD, HS, PS, and RS) chronic skin diseases
The correlation-based gene clustering is used to reduce the high-dimensional transcriptomics datasets for easier human interpretations by clustering genes into several groups based on their expression profile. GCN-based clustering is also a subtype of conventional clustering with reduced complexity through network representation. The resultant GCN can be a source point to predict the relationships among genes in a biological pathway through the guilt-by-association concept27. However, the main challenge is to restrict spurious correlations, which can be achieved to a great extent by using datasets with a higher number of samples/conditions, robust GCN network construction algorithms, and standard parameters for each experiment. Thus, we performed our correlation-based analysis through WGCNA with the same parameters of power-cutoff, weight-cutoff, and clustering algorithm for eight inflammatory skin disease transcriptomes23. There are eight resultant GCNs of which acne (320 nodes and 264 edges) is the smallest, HS is the largest by an edge (4162 nodes and 420,422 edges) and, PS (5465 nodes and 132,849 edges) is the largest by nodes. AK, CD, and ICD have a comparable number of nodes and an edge. (Fig. 3A and Data S2).
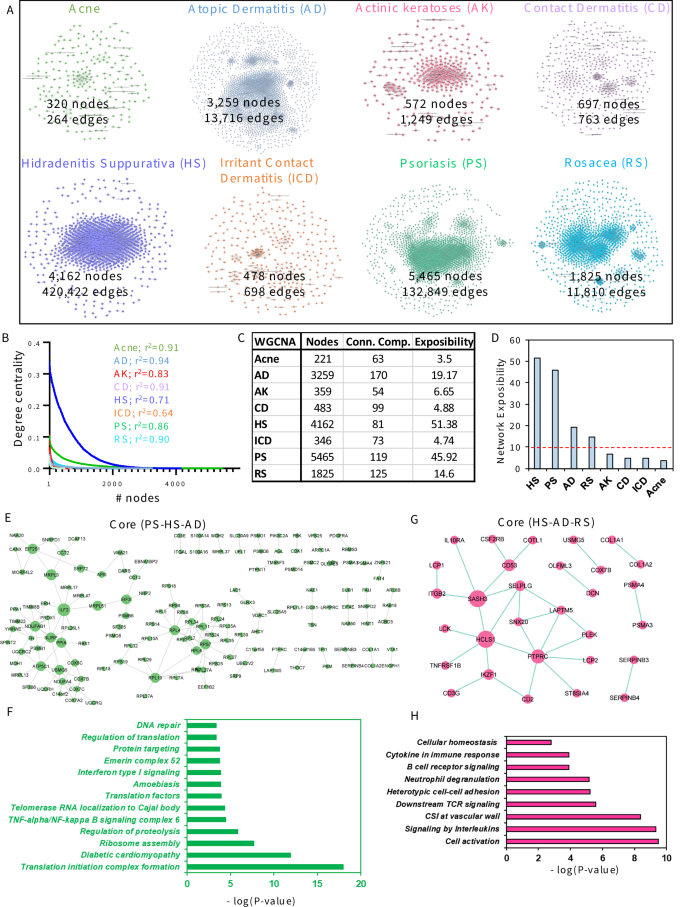
A Different co-expression networks were constructed with the same parameters through WGCNA for eight inflammatory skin diseases with the corresponding number of nodes and edges. The networks are acne (320 nodes and 264 edges), atopic dermatitis (AD; 3259 nodes and 13,716 edges), actinic keratoses (AK; 572 nodes and 1249 edges), contact dermatitis (CD; 697 nodes and 763 edges), hidradenitis suppurativa (HS; 4162 nodes and 420,422 edges), irritant contact dermatitis (ICD; 478 nodes and 698 edges), psoriasis (PS; 5465 nodes and 132,849 edges), and rosacea (RS; 1825 nodes and 11,810 edges). B The power-law distribution of eight co-expression networks to measure their scale-freeness. C The table illustrates the total number of nodes and connected components to determine the network exposibility for each co-expression network. D The network exposibility distribution for eight co-expression networks to illustrate the robust networks to study further with parameter (network exposibility > 10). E The core co-expression network among psoriasis, hidradenitis suppurativa, and atopic dermatitis. F The gene ontology analysis of enriched pathway of core co-expression network genes among PS, HS, and AD (P-value < 0.05). G The core co-expression network among HS, AD, and RS. H The gene ontology analysis of enriched pathway of core co-expression network genes among hidradenitis suppurativa, atopic dermatitis, and rosacea (P-value < 0.05).
To begin our GCN analysis, we performed several network centrality analyses including the degree to check the scale-free properties of each network28. As expected, we found that all eight GCNs; Acne, AD, AK, CD, HS, ICD, PS, and RS follow scale-free properties calculated by the power-law distribution with r2 values 0.94 for AD and 0.64 for ICD (Fig. 3B and Data S2). Though most of these GCNs are biologically relevant through power-law distribution, they don’t seem comparable to each other. Therefore, random network analysis was performed and plotted on all these networks to further explore the key features. From the results, we found all eight GCNs showed relatively high neighborhood connectivity, especially for PS, ICD, and AD, which are 0.5164, 0.4764, and 0.419 (Fig. S2B), and significantly high clustering coefficients as 0.7159 for PS that indicates the co-expression networks have strong local interactions. To check the extent of coverage in a network, we calculated the network exposibility as described in the methods section. We found that four GCNs corresponding to acne, AK, CD, and ICD have a small number of nodes and a high number of connected components, whereas the remaining four other GCNs related to AD, HS, PS, and RS have a high number of nodes but a smaller number of connected components comparably (Fig. 3C and Data S2). To determine the exposibility-based reliability of eight GCNs, we set a network exposibility threshold ≥ 10 and filtered our four disease-specific GCNs including HS, PS, AD, and RS for further analysis (Fig. 3D).
It is well reported that some of the immune-mediated diseases including PS, HS, and AD share most of their disease-associated pathways and comorbidities with each other29,30. Therefore, to understand the shared gene patterns among these diseases, we explored the core co-expression network of four diseases. Interestingly, we report a core co-expression network among PS, HS, and AD with 145 genes and 161 associations (Fig. 3E and Data S2). Interestingly, some of these genes are RPS-, RPL-, MRPL-, EIF-, COX-family genes, ILF2, PPIA, NDUFAB1, NDUFA4, ATP5C1, and FH. Furthermore, to verify the biological associations of 145 core genes, we performed gene ontology analysis. The ontology analysis identified the enriched pathways including translation initiation complex formation, diabetic cardiomyopathy, ribosome assembly, regulation of proteolysis, TNF-alpha/NF-kappa B signaling complex 6, telomerase RNA localization to Cajal body, translation factors, amebiasis, Interferon type I signaling, Emerin complex-52, protein targeting, and DNA repair31,32,33,34,35 (P-value < 0.05; Fig. 3F and Data S2).
Similarly, we explored the core co-expression network among PS, HS, and RS. In this analysis, we found a small core network encompassing 30 genes and 30 associations (Fig. 3G and Data S2). Interestingly, some of these genes are IKAROS Family Zinc Finger 1 (IKZF1), CD2 molecule (CD2), CD53 molecule (CD53), SAM And SH3 domain containing 3 (SASH3), Lysosomal Protein Transmembrane 5 (LAPTM5), Interleukin 10 Receptor Subunit Alpha (IL10RA), Protein Tyrosine Phosphatase Receptor Type C (PTPRC), and Pleckstrin (PLEK). Furthermore, to verify the biological associations of 30 core genes in HS, AD, and RS, we performed gene ontology analysis. We found that most of the enriched pathways are cell activation, signaling by Interleukins, CSI at the vascular wall, downstream TCR signaling, heterotypic cell-cell adhesion, neutrophil degranulation, B cell receptor signaling, cytokine in immune response, and cellular homeostasis33,35,36,37,38,39,40,41 (P-value < 0.05; Fig. 3H and Data S2). These results suggest the significance of correlation-based GCN construction and analyses to identify the emerging and shared players in associated pathways among inflammatory skin diseases.
Interactome analysis identified the central proteins in psoriasis, hidradenitis suppurativa, atopic dermatitis, and rosacea
The molecular mechanisms and intricacies of biological processes associated with immune-mediated skin diseases are mostly determined by protein interactions in certain pathways42. Given that the coverage of DEGs from transcriptome to differentially expressed proteins is a maximum of only ~60%, we used the disease-specific co-expressed gene list to extract the PPI network. These interactions can be more relevant in any stress condition if they are also part of corresponding GCNs by satisfying the guilt-by-association assumption43. To explore these possibilities, we constricted the disease-specific PPI network by integrating the co-expressed genes into the known interactions from the STRING database25. The resultant PPI networks/interactomes for these four diseases include PS with the greatest number of nodes (3622 nodes and 33,173 edges) and RS with the least number of nodes (851 nodes and 3234 edges) (Fig. 4A and Data S3). It is reported that some of the molecular pathways and disease comorbidities are shared among PS, HS, AD, and RS1. To identify the core proteins in four PPIs, we performed the commonality analysis and found that 53 proteins are shared among these four inflammatory skin diseases (Fig. 4B and Data S3). Some of these proteins are CD2, CD3D, CD53, CD96, CDK1, COL1A1, COL1A2, COL3A1, COX5A, COX7B, CXCR4, DCN, FH, IKZF3, IL10RA, ITGAL, KRT6A, KRT6B, LAPTM5, and LCK, which participate in immunodeficiency, anemia, Th1 and Th2 cell differentiation, Th17 cell differentiation, lymphocyte activation, diabetic cardiomyopathy, neutrophil degranulation, membrane raft distribution, lymphocyte-mediated immunity, TYROBP causal network in microglia, response to molecule of bacterial origin, microglia pathogen phagocytosis pathway, homeostasis of several cells, formation of the cornified envelope, negative regulation of defense response, cell cycle phase transition, positive regulation of endopeptidase activity, regulation of glial cell differentiation, acute viral myocarditis, human cytomegalovirus infection, and immune effector process (Figs. S3 and S4 and Data S3).
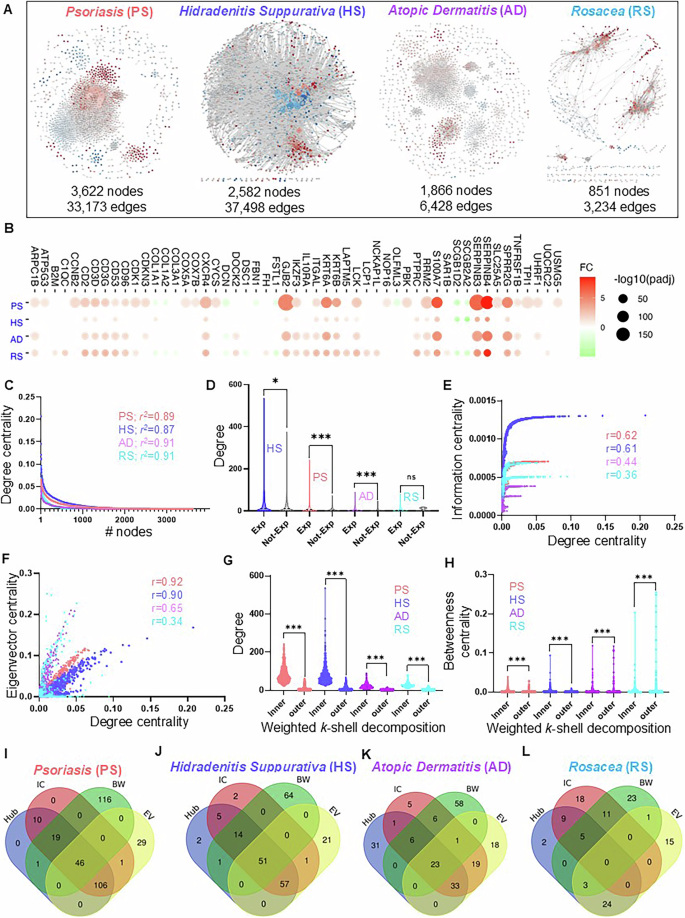
A Different PPI networks were constructed with a compendium of human interactome extracted by co-expression network nodes for four inflammatory skin diseases with a corresponding number of nodes and edges. The PPI networks are psoriasis (PS; 3622 nodes and 33,173 edges), hidradenitis suppurativa (HS; 2582 nodes and 37,498 edges), atopic dermatitis (AD; 1866 nodes and 6428 edges), and rosacea (RS; 851 nodes and 3234 edges). B The shared proteins in all four PPI and their expression pattern across four diseases. Upregulated DEGs are marked in light red and down-regulated DEGs are marked in light green. C The power-law distribution calculation of eight co-expression networks to measure their scale-freeness. D The degree distribution of expressed genes (Exp) and not expressed genes (Not-Exp) in four disease-specific PPIs. E, F The correlation plots illustrate the distribution among degree, information, and eigenvector of all four PPI networks. G, H The distribution of inner and outer layer proteins with a degree, as well as betweenness centrality in all four PPI networks. I–L The shared and unique proteins with significantly high centralities (top 5%) for individual disease-specific PPI networks.
The disease-specific interactomes position their important proteins in a particular arrangement to communicate throughout the network most efficiently. To explore these structural arrangements, we performed network analysis on four disease-specific PPIs including PS, HS, AD, and RS. To begin our analysis first, we tested the biological relevance of interactomes by performing the power-law distribution analysis of four PPIs to verify their scale-freeness28,44. Interestingly, we report that all four PPIs for PS, HS, AD, and RS follow scale-free properties calculated by the power-law distribution and the r2 values are similar (Fig. 4C and Data S3). Random network analysis was also applied to all PPI networks to further explore their features. The results showed all four PPI networks for PS, HS, AD, and RS showed an obviously high clustering coefficient which are 0.4945, 0.4586, 0.4814, and 0.4479 which indicates they have strong local interactions (Fig. S3A). Furthermore, the high neighborhood connectivity also proves there are strong local interactions in these four PPI networks as the values of 0.4408, 0.3638, 0.3333, and 0.3303 for PS, HS, AD, and RS. Previously it has been reported that the network-based method has successfully highlighted the central and core proteins associated with disease pathogenesis13,42,44. Therefore, we leveraged a part of this analysis in our framework and analyzed four PPIs with a degree, betweenness centrality, eigenvector, information centrality, and weighted k-sell decomposition method. First, we explored the degree distribution of expressed genes (Exp; FDR < 0.05) and non-expressed (Not-Exp; FDR > 0.05) for each disease. Interestingly, we found that expressed genes of all four diseases namely HS, PS, AD, and RS encompass a high degree distribution than their non-expressed counterparts (Fig. 4D and Data S3), Mann–Whitney test P-value (HS < 0.05, PS < 0.0001, AD < 0.0001, and RS > 0.66).
Previously, we have demonstrated that some of the highly connected genes also possess other network properties, which make them extremely vulnerable during disease pathogenesis13,22. Therefore, we calculated the correlation between degree centrality and other centralities including information, eigenvector, and betweenness centralities of all four PPI networks. Interestingly, we found that the degree of PS and HS are strongly correlated with information centrality with r > 0.6 for both, whereas this correlation is not strong in AD and RS with r < 0.5 (Fig. 4E). Similarly, we computed the correlation among degree and eigenvector centrality and reported that PS, HS, and AD have the strongest correlation (r < 0.5), whereas RS has the weakest correlation (r < 0.5, Fig. 4F). Differently, the correlation between degree and betweenness is very poor in PS, AD, and RS (r < 0.5) and strong in HS (r > 0.5) (Fig. S3B). Further, we decomposed the PPI networks into k-shells through the Weighted k-shell decomposition method to identify the hidden genes, which the conventional centralities fail to identify17. Next, we identified the inner layer and peripheral (outer) layer proteins in each PPI as mentioned by Ahmed et al.17. We report that there are 461, 573, 393, and 95 proteins in the inner layer and 3135, 1982, 1466, and 754 proteins in the outer layer of PS, HS, AD, and RS, respectively (Data S3). Moving forward, we explored the degree and betweenness centrality distributions of the inner layer and outer layer proteins in four PPIs. We report that inner-layer proteins possess a significantly higher degree compared to outer-layer proteins (Fig. 4G; Mann–Whitney test P-value < 0.0001, Data S3). However, we do not see much difference in the betweenness centrality distribution of inner and outer layer proteins (Fig. 4H; Mann–Whitney test P-value < 0.0001). These analyses confirm that some of the proteins of PPIs share different types of high centrality values, which can be central in the disease pathogenesis. Additionally, we investigated the high centrality proteins (significant proteins) by selecting the top 5% of high centralities i.e Hub (degree), bottleneck (Betweenness, BW), information centrality (IC), and eigenvector centrality (EV) for individual disease-specific PPI network. We demonstrate that 46, 51, and 23 proteins are shared by all four centralities in PS, HS, and AD, respectively (Fig. 4I–K and Data S3). Whereas there was not a single protein shared by all four centralities in RS (Fig. 4L Data S3). Interestingly, we also found that most of the high betweenness centrality (bottleneck) proteins are not shared by any other centrality. These analyses indicate that most of these bottlenecks are part of smaller subnetworks in four PPIs.
Network centrality-based prioritization of proteins and pathways in chronic inflammatory skin diseases
It has been previously described in several instances that in the disease-specific interactomes, the most significant contributing proteins have a high degree (connections) and high betweenness (bottlenecks) against other proteins throughout the PPI network18,19,20,42,45. Further, the analysis was expanded to other centralities including information and eigenvector centralities, which improved the identification of significantly contributing proteins in the interactome13. Inspired by these studies, we identified the significant proteins for disease-specific PPI networks of PS. HS, AD, and RS individually. The top 5% of centrality value nodes with the degree, betweenness, eigenvector, information centrality, and the inner layer proteins from the weighted k-shell decomposition method were identified as significant proteins. Next, we put effort into classifying these significant proteins as regulators in four skin diseases in this study and termed them as HPPs or significant regulators (Fig. 5A and Data S4). As a result, we identified 55 HPPs that contribute significantly to the pathogenesis of four selected diseases (Data S4). Further, we explored the regulator activity of these 55 HPPs and found that most of these proteins are significantly activated in at least seven inflammatory skin diseases (Fig. 5B, BH P-value < 0.05; Activation z-score > |1|; Data S4). Interestingly, some of these 55 HPPs are involved in RTKs signaling, proteoglycans in cancer, cell morphogenesis, hemopoiesis, PID CXCR4 pathway, epithelial cell differentiation, cytokine signaling in the Immune system, adherens junction, regulation of kinase activity, cellular response to lipid, head, and neck SCC, response to wounding, Hippo signaling regulation46, Interferon type I signaling, PI3K-Akt signaling, leukocyte activation, response to estradiol, lymphocyte activation37, Th1, and Th2 cell differentiation39, and NK cell-mediated cytotoxicity41 (Fig. 5C, Data S4; P-value < 0.05). Most of these HPPs and associated pathways are known for their major contribution to several immune-mediated diseases5,10,11,42,44,47,48,49,50. Additionally, we performed disease association experiments using GWAS databases including DisGeNET and enrichR disease ontology. Most of the highly enriched diseases for HPPs are cancer, IBD, MS, Colitis, Lupus, and other inflammatory diseases (Fig. S5). Important to this study, hubs including STAT1, TP63, CD2, EGFR, and YAP1 have been previously reported in multiple skin conditions including HS, and could be promising targets for broader therapeutic applications41,51,52. Recently, we have identified CD2 as an important therapeutic target and utilized an anti-CD2 therapy to reduce the cytokine/chemokine production in Hidradenitis suppurativa in organoids culture41. We also evaluated the enrichment of IKZF1 in negative control ARCHS4 database53 and found its enrichment in immune (T and B) cell activation and differentiation with fair expression in Skin tissue (Fig. S6). Some of the mouse phenotypes associated with IKZF1 are abnormal thymus physiology, abnormal humoral immune, abnormal bone marrow, and B cell deficiency. These results highlight the significance of these hubs in the pathogenesis of different skin diseases.
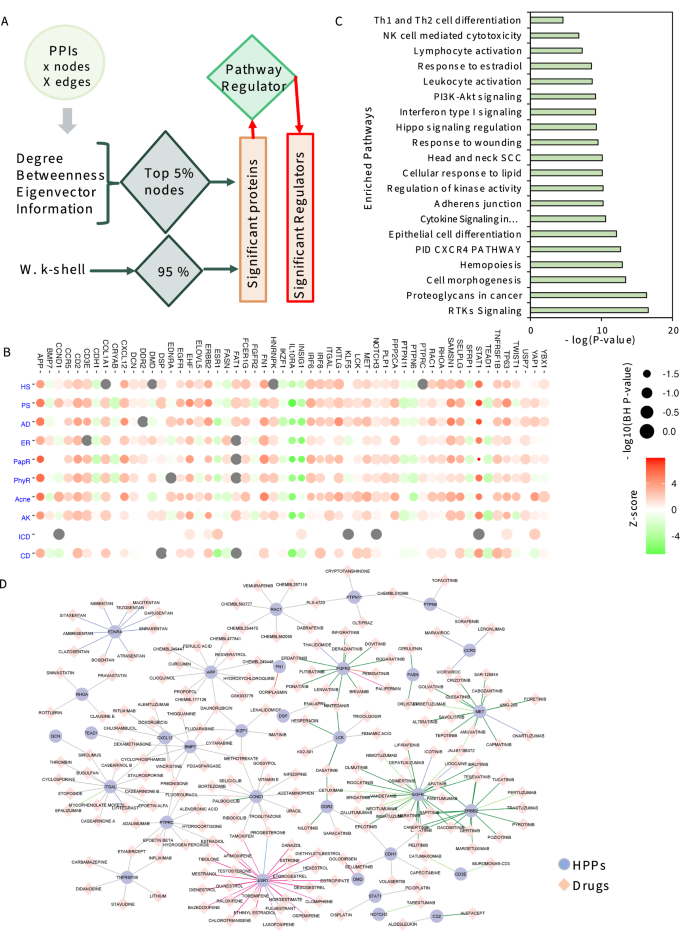
A The framework to identify the HPPs for each PPI network. The top 5% of centrality value nodes with a degree, betweenness, eigenvector, information centrality, and the inner layer proteins from the weighted k-shell decomposition method were identified as significant proteins. If these significant proteins are also significantly activated or inhibited regulators in each of the eight inflammatory skin diseases are termed HPPs or significant regulators. B Our network-centric approach identified 55 HPPs and their activity across inflammatory skin diseases. The activated regulators are marked in light red and the inhibited regulators are marked in light green. C The gene ontology analysis of the enriched pathway of 55 HPPs (P-value < 0.05). D Drug–gene interaction network pairs for HPPs and chemical compounds with publication listed in DGIdb.
Subsequently, we investigated the reported potential therapeutics for candidate 55 HPPs, which may help in designing highly effective drug repurposing strategies. We extracted the drug–gene interaction network pairs from DGIdb26 for 55 HPPs with available literature publications. We found a total of 32 HPPs have a known compendium of 199 drug compounds with 237 interactions (Fig. 5D and Data S4). Interestingly, we report that some 55 HPPs i.e., CD2, LCK, STAT1, TNFRSF1B, IKZF1, APP, and BMP7 can be a high-priority drug target for several of these chronic skin diseases. Recently, we described the role of CD2 in the molecular pathogenesis of HS. We specifically defined the role of NK, NKT, and T cells in the disease progression of HS41.
IKZF1 is a potential therapeutic target for several inflammatory skin diseases
Given the overlap in the molecular profiles of multiple inflammatory skin diseases, we explored the shared significant regulator proteins in HS, PS, AD, and RS. For this, we complied all the significant proteins based on five centrality methods (degree, betweenness, information, eigenvector centrality, and weighted k-shell decomposition) for each of these diseases. As a result, we have identified a list of 236, 195, 168, and 72 significant proteins, respectively in PS, HS, AD, and RS (Fig. 6A and Data S4). Afterward, we studied the distribution of shared and unique significant proteins in each PPI network and found that six proteins (NDUFAB1, MRPL3, DDX1, EIF2S1, SLIRP, and ATP5C1) are contributing significantly to these four diseases. Interestingly, most of these genes are related to mitochondrial metabolism, protein translation in DNA damage. Earlier DNA damage and mitochondrial damage-related signaling were found to be associated with multiple diseases including HS in the skin34,54. In addition, these genes have also been associated with HS-related co-morbidities including heart and brain comorbidities55,56,57,58. Whereas there is only one protein IKZF1 contributing significantly to HS, AD, and RS with high network centrality values, we explored the drug-gene interaction of IKZF1. In this regard, six drugs including lenalidomide, daunorubicin, methotrexate, cytarabine, imatinib, and fludarabine are known to target IKZF1 (Fig. 6B). Some of the drugs that we identified have been reported to alter IKZF1 function and are a part in clinical trials. In fact, lenalidomide alone and in combination with other drugs including pomalidomide have been reported to manage multiple myeloma and limit autoimmunity and immune cell activity59,60,61. Interestingly, we also found that daunorubicin also targets APP and BMP7 which are also part of predicted 55 HPPs. Similarly, methotrexate also targets CCND1 and BMP7, and cytarabine targets BMP7 too (Fig. 5D). Thereafter, we extracted all the possible interactions of IKZF1 from HS-PPI, HS-GCN, and known TF-target relationships to explore the extrapolating effect of a drug on the IKZF1 and its partners. We found that IKZF1 can be a really good candidate as it acts as a master regulator by interacting with 19 targets, 62 proteins, and 88 co-expressed genes (Fig. 6C and Data S4). We hypothesized that the activities of these proteins and co-expressed genes can be altered by targeting IKZF1 with a specific drug, such as lenalidomide. As IKZF1 is shared among three of these skin diseases, we surveyed the expression pattern of IKZF1 interacting and co-expressing genes in HS, PS, AD, and RS. Interestingly, we found that most of the IKZF1 interacting partners; 19 targets, 62 proteins, and 88 co-expressed genes follow a similar expression pattern across four inflammatory skin diseases (Fig. 6 D–F and Data S4). This is consistent with our own observations demonstrating some efficacy of lenalidomide in suppressing inflammatory responses in our HS model7.
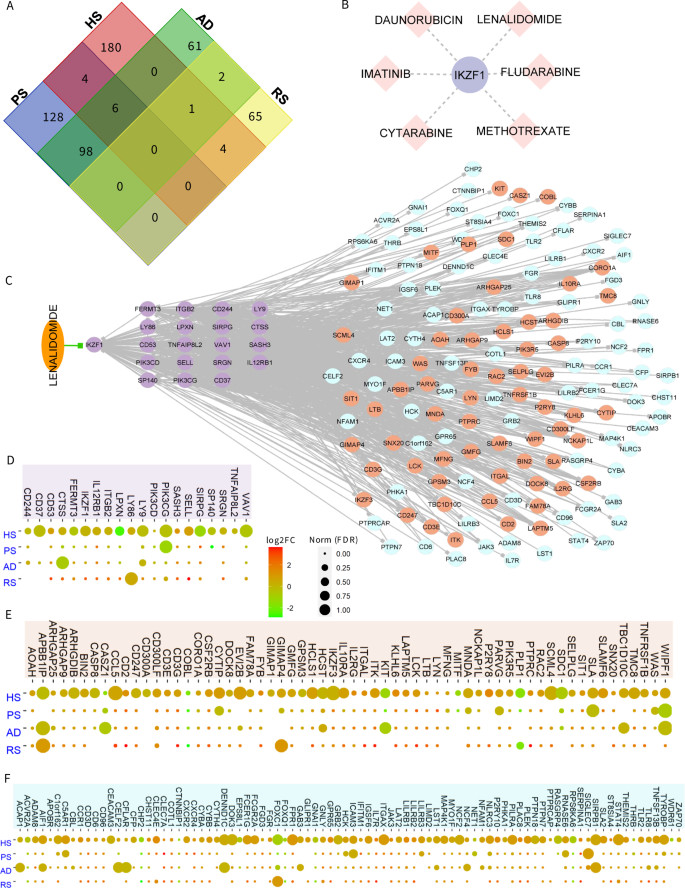
A The Venn diagram represents the distribution of shared and unique significant proteins in each PPI network. Interestingly, IKZF1 protein is shared among hidradenitis suppurativa (HS), atopic dermatitis (AD), and rosacea (RS) with high network centrality values. B The six drugs are known to target IKZF1. C The IKZF1 acts as a master regulator by interacting with 19 targets, 62 proteins, and 88 co-expressed genes. Activities of these proteins can be altered by targeting IKZF1 with the drug Lenalidomide. D–F The expression profile of IKZF1 interacting partners; 19 targets, 62 proteins, and 88 co-expressed genes in four inflammatory skin diseases including HS, PS, AD, and RS.
Taken together, our study identified high-value proteins involved in the pathogenesis of chronic skin diseases including HS, PS, AD, and RS. Additionally, we designed a robust framework to identify significant contributors utilizing transcriptome datasets and integrative multi-omics approaches supported by network systems biology for more accurate predictions of regulators in most chronic skin diseases.
Discussion
Almost 1/5th of the human population across the world is affected by some type of non-communicable chronic skin inflammatory disease62. The disease manifestation and immune regulatory signatures are shared among some of these inflammatory skin diseases. The high-throughput ‘omics technology and robust multi-omics network integration techniques showed immense potential to accelerate more comprehensive and system-wide discoveries in the molecular pathogenesis of complex inflammatory skin diseases. Here, our designed robust framework, identified significant contributors from different transcriptome datasets and integrative multi-omics supported the network systems biology for more accurate predictions of significant regulators of these inflammatory skin diseases (namely: acne, AD, AK, CD, ICD, PS, HS, and three types of RS). Our conventional and unconventional biological data analysis approach extracted the shared and unique biomolecular (gene/protein/regulator) signatures and biological processes in four (PS, PS, AD, and RS) of these eight inflammatory skin diseases. Further, systems genetics and network biology-driven analyses were instrumental in prioritizing the most relevant proteins (55 high-priority proteins; HPPs) across four of these inflammatory skin diseases. These HPPs can serve as a template to study the impact and response of disease pathogenesis of these inflammatory skin diseases, and putative drug targets strategies either as standalone or combinatorial approaches. Overall, this study employs advanced and powerful network systems-based analyses that combine integrative multi-omics to establish a mutual understanding of inflammatory mechanisms in PS, PS, AD, and RS.
In the last two decades, PS has been one of the most studied inflammatory skin diseases63, which shares a significant number of immune-mediated pathways with other inflammatory diseases including acne, AD, AK, HS, and RS. For example, similar to psoriasis, targeting Th2 cells, IL-4, and IL-13 have been the most efficacious (50–70%), and advanced therapy for AD49 and Janus kinase (JAK) inhibitors has been demonstrated to suppress the cytokine responses more effectively and are also a powerful treatment strategy for AD49. HS is also an inflammatory skin disease implicated by the pathogenesis of neutrophilic inflammation, dysbiosis, TNF, interferon responses, hair- and skin-gland abnormalities, autoantibodies, and plasma cells50. Our multi-omics analysis identified the shared molecules, regulators, and pathways at transcriptome and interactome layers in four diseases including TNF-alpha/NF-kappa B signaling, translation factors, amebiasis, Interferon type I signaling, Cytokine signaling, and interleukin signaling. These pathways have been under investigation to manage/treat several immune system diseases35,64. For example, our group has reported evidence of dysregulated protein translation control through upregulation in expression and phosphorylation of eIF4E in HS skin31. Aberrant expression and activation of serine proteases and PAR-2 have been observed in AD lesional skin32. TNF-α is a key inflammatory player in HS, PS, and AD32,33,35. Anti-TNF therapies like adalimumab have shown efficacy in treating HS35. Similarly, type I interferon signatures are reported with the HS disease34. Immune cell activation in HS, AD, and RS36,39,41, signaling by Interleukin35,38, and B cell receptor signaling in HS65. Taken together, our analysis framework identified the most intrinsic properties of skin diseases.
Our network analytics utilizing different conventional and improved centralities appreciably identified the central proteins of these skin disease-specific interactomes, as well as substantially improving the classification of significant 55 HPPs in the pathogenesis of PS, HS, AD, and RS. The functional analysis of these HPPs further highlighted the Cytokine Signaling, adherens junction, response to wounding, Hippo signaling regulation, Interferon type I signaling, PI3K-Akt signaling, leukocyte activation, Th1, and Th2 cell differentiation, NK cell-mediated cytotoxicity to name a few. Many of these pathways have been validated in various earlier investigations42,49,66. Recently, we have demonstrated the involvement of many of these cellular and molecular signaling pathways in the pathogenesis of HS41, which is the current focus of our laboratory. These findings demonstrate the effectiveness of a network centrality-based framework to untangle the underlying players of these diseases.
Our integrative approach combines network topology with multi-omics data, leveraging five distinct centrality measures—degree, betweenness, eigenvector, information centrality, and weighted k-shell decomposition—to uncover key regulatory proteins that may act as “master regulators.” By expanding beyond traditional methods and incorporating diverse centrality metrics, our strategy reveals hidden patterns and regulatory mechanisms, offering novel insights and potential advancements in therapeutic and biotechnological applications (Fig. 5). Recent studies have demonstrated the significant impact of modeling regulatory network topology on understanding the genetic basis of complex traits67,68,69. By analyzing 18 human traits across 38 distinct regulatory networks, it was shown that genetic signals associated with these traits are frequently enriched within interconnections that are specific to the cell types or tissues most relevant to the traits67. This finding highlights the importance of context-specific regulatory interactions in elucidating the underlying genetic mechanisms. Furthermore, heritability enrichment within these context-specific regulatory networks has been shown to enhance the identification of phenotype-relevant tissues, offering a more precise approach to linking genetic variations with their functional consequences in specific biological contexts68. These insights highlight the potential of leveraging network-based methodologies to deepen our understanding of trait-specific genetic architecture.
Our integrative multi-omics framework has identified IKZF1 as a significant shared regulator among HS, AD, and RS. In this study, IKZF1 is associated with chromatin remodeling and lymphocyte differentiation which can influence the disease pathogenesis. The diverse mechanisms linked to IKZF1 dysfunction contribute to a spectrum of immune-related conditions, including heightened vulnerability to infections, autoimmune diseases, allergic reactions, and various types of cancer. These alterations in IKZF1 function can disrupt normal immune responses, leading to a range of pathological outcomes that span from impaired pathogen defense to aberrant immune activation and malignant transformation70. Of note, several studies have already established IKZF1’s significant role in various autoimmune and inflammatory conditions. Hu et al.71 demonstrated IKZF1’s critical function in systemic lupus erythematosus pathogenesis71. Klarić et al. (2020) revealed IKZF1’s involvement in regulating IgG glycosylation, which is relevant to multiple autoimmune diseases72. Hoshino et al.73 identified IKZF1 variants impacting the lymphoid differentiation stages which are associated with a wide range of inflammatory, autoimmune, and allergic symptoms73. More recently, Garcia-Solis et al.74 reported on IKZF1 gain-of-function variants causing IgG4-related disease and B-cell malignancy74. IKZF1 also serves as a regulator of immune infiltration in solid tumors and therefore provides a molecular therapeutic target for enhancing susceptibility to immunotherapy75.
A considerable amount of IKZF1 interacting partners in HS interactome and co-expression networks suggest the involvement of this transcription factor in the immune environment modulation in the disease pathogenesis. This transcription factor has earlier been identified as a master regulator together with DLX4, which underpins the pathogenesis of Alopecia Areata. In this process, IKZF1 and DLX4 induce Alopecia Areata-like immune infiltration76.
Interestingly, we also found most immune-system pathways including cellular response to cytokine stimulus, leukocyte activation, neutrophil degranulation, inflammatory response, TCR pathway, CXCR4 pathway, hemostasis, and microglia pathogen phagocytosis pathway enriched among IKZF1 interactors. The specific role of neutrophils, macrophages, B cells, and plasma cells has been identified in HS pathogenesis65,66,77. Similarly, CXCR4 pathways are considered important in regulating B cell functions, homing of plasma cells, and the migration of T cells78,79. Interestingly, many small molecule drugs are known modulators of IKZF1 responses. Among these, multiple immunomodulatory drugs are being contemplated for various skin diseases including those of HS. In our ex vivo skin culture models we showed some efficacy of lenalidomide in suppressing cytokine chemokine production7. It is likely that these or similar molecules may find a place in developing therapeutic innervations of these complex inflammatory skin diseases. We highlighted IKZF because it has been found in HS and can contribute to its pathogenesis. However, we anticipate that IKZF1 alone might not be highly effective in completely abrogating the HS symptoms. Therefore, additional molecular targets including transcription targets should be investigated to develop combination therapy that may manifest highly efficacious response80,81. For instance, here we identified additional potential targets in concert with IKZF1, which may be playing a critical role in the pathogenesis of HS and other similar chronic inflammatory skin diseases. STAT1 is involved in interferon-gamma signaling, which has both pro-inflammatory and pro-repair functions in intestinal inflammation81. TNFRSF1B (also known as TNFR2) is part of the TNF signaling pathway, and targeting the TNF/TNFR superfamily has shown success in treating various immune-mediated inflammatory diseases80. EGFR signaling has been linked to inflammatory processes, and its inhibition can reduce STAT1 levels, potentially modulating immune responses82. Recently, we demonstrated the role of CD2 targeting in attenuating inflammatory response in HS41. Combining targets, such as IKZF1 with CD2, STAT1 with TNFRSF1B, or EGFR with STAT1, could offer synergistic effects in modulating inflammatory responses. These regulators are now reported by several high-throughput single-cell and spatial transcriptome experiments and thus could be easily employed as a testable hypothesis. Importantly, further research is needed to fully understand their interactions while developing effective therapies and uncover the molecular basis of adverse effects. Overall, our integrative network-based multi-omics framework identified the underlying regulators, and pathways common among these diverse proinflammatory diseases.
This study also has limitations as available datasets for bulk transcriptome-driven/disease integrative multi-omics inflammatory skin diseases, will not generate the most reliable prediction of significant regulators, yet this framework as suggested here can be applied for multiple transcriptome datasets of a single disease for more accurate predictions. Furthermore, expanding the current version of transcriptome-driven integrative systems to the next level by implementing other multi-layered datasets including systems genomics, epigenomics, single-cell RNA sequencing, and spatial transcriptomics will uncover the high-resolution molecular pathogenesis, pathways, and cell-cell communications in chronic inflammatory skin disease.
Methods
Data acquisition
Publicly available transcriptome datasets were extracted from NCBI GEO in eight different inflammatory skin diseases including hidradenitis suppurativa (HS, GSE72702)83, rosacea (RS, GSE65914)48, atopic dermatitis (AD, GSE121212)84, contact dermatitis (CD, GSE6281)85, actinic keratoses (AK, GSE90643)86, Irritant contact dermatitis (ICD, GSE18206)87, Acne (GSE53795)47, and psoriasis (PS, GSE121212)84.
Transcriptomics analysis
The differentially expressed genes (DEGs) analysis was performed on all eight disease datasets with the same threshold parameters24. DESeq2 was used for RNA-Seq experiments and edgeR was used for microarray experiments. Standard cutoff i.e. log2fc ≥ |1| and adjusted P-value by FDR < 0.05 was selected for DEGs quantification.
Co-expression network constriction
We performed the gene co-expression network (GCN) construction and analysis for all eight disease datasets through weighted gene co-expression network analysis WGCNA23. First, we preprocess the gene expression data by removing low-expression genes and outliers. Next, we construct a similarity matrix based on pairwise correlations between genes and transform it into an adjacency matrix using a soft-thresholding power. This adjacency matrix is then used to calculate topological overlap, a robust measure of interconnectedness. We perform hierarchical clustering based on topological overlap to identify modules of co-expressed genes. Finally, for network topological analyses and visualization, the weighted undirected co-expression network was extracted utilizing a hard threshold of weight ≥ 0.75 and exported into Cytoscape v.3.7.1 by exportNetworkToCytoscape function. We achieved eight disease-specific networks ranging from 320 nodes with 264 edges in Acne to 5,465 nodes with 132,849 edges in PS. This analysis demonstrated the challenges in the handling of multivariate transcriptomes. Networks were visualized in Cytoscape88.
Protein–protein interaction network construction
To establish the disease-specific protein–protein interaction network we fused the results from co-expression networks, DEGs, and the largest publicly available human protein–protein interaction network from the STRING database25. Finally, we had a collection of eight disease-specific protein–protein interaction networks ranging from 90 nodes with 90 edges in Acne to 3622 nodes with 33,173 edges in PS. Next, we performed network centrality analyses on all four filtered networks to identify the most important players in each disease13. We also computed the inner core proteins identification by weighted k-sell decomposition method17. Networks were visualized in Cytoscape88.
Random network generation
10 random networks were generated for each original network (both co-expression networks and PPI networks) through expected_degree_graph function from networkx 3.4.2. Each random network was analyzed by Network Randomizer 1.1.3 under Cytoscape 3.7.2 compared to the original network.
Network analysis
NetworkX and Cytoscape were utilized to calculate network centralities of gene co-expression and protein–protein interaction networks as a graph. The graph is represented through nodes (genes/proteins) and edges (connections) based on the respective relationships. We computed the network measures covering neighborhood-based, path-based, and iterative refinement centralities. For example, degree Centrality measures the number of direct connections a node has. In a co-expression network, a high degree of centrality indicates a gene that is co-expressed with numerous other genes, suggesting a central role in the network. The degree_centrality() function from NetworkX was used to calculate the degree centrality. Betweenness Centrality quantifies how often a node acts as a bridge along the shortest paths between other nodes. Genes with high betweenness centrality might be crucial for connecting different functional modules in the network. The betweenness_centrality() function was used for this calculation. Information Centrality assesses a node’s importance based on its contribution to the overall information flow in the network. Genes with high information centrality are considered vital for transmitting information across the network. The information_centrality() function is employed here. Eigenvector Centrality assigns relative scores to nodes based on the centrality of their neighbors. A high eigenvector centrality indicates a gene that is connected to other highly connected genes, highlighting its influence in the network. The eigenvector_centrality() function was utilized for this calculation.
Network exposibility analysis
To check the extent of coverage in a network and inspired by the COVID-19 exposibility scenario89, we calculated the network exposibility by dividing the total number of nodes by the total connected components in the network. Based on the network exposibility threshold ≥ 10 to determine the reliability of eight PPI networks, we filtered four disease-specific interaction networks including HS, PS, AD, and RS for further analysis.
High-priority protein identification
To identify the High-priority proteins (HPPs) or significant proteins for each PPI network, we calculated the top 5% of centrality value nodes with the degree, betweenness, eigenvector, information centrality, and the inner layer proteins from the weighted k-shell decomposition method. Next, if these significant proteins are also significantly activated or inhibited regulators characterized by pathway analysis in each of the eight inflammatory skin diseases, then these proteins are termed HPPs or significant regulators. Additionally, we identified the most important regulators shared among four diseases and mapped them against their regulated pathways.
Drug-gene interaction network
We extracted the HPPs interacting drugs from the publicly available databases DGIdb26. Only the interactions with listed publication information were included in this analysis. These drug-gene (HPPs) interactions can provide significant information about the potential therapeutic options for a disease or class of diseases. The drug-gene interaction network was visualized in Cytoscape88.
Gene ontology and pathway analysis
The canonical pathway and regulator analysis was performed by IPA with default parameters. The gene ontology and pathway analysis were performed through metascape90 with standard parameters.
Significance analysis
The DEG analysis was performed with (log2FC ≥ |1|; FDR < 0.05) parameters. The canonical pathway analysis was performed with a significance test BH corrected P-value < 0.05. The gene ontology analysis was performed with a significance test P-value < 0.05. The network power-law distribution fitness threshold is r2 ≥ 0.5. The network exposibility threshold was 10 for reliable networks. The correlation among values of different centralities is positive if r ≥ 0.5. The significance of degree and betweenness among the inner and outer layers of four PPIs were tested by the Mann–Whitney test for non-parametric distributed values. The significant regulator analysis through IPA was tested by BH P-value < 0.05.
Responses