Lessons learned from a candidate gene study investigating aromatase inhibitor treatment outcome in breast cancer

Introduction
Breast cancer is the most frequent cancer and leading cause of cancer death in women1 affecting both pre- and postmenopausal women. Most breast cancers (75%) express the estrogen receptor α (ERα) known to fuel tumor growth through estrogen signaling. Its presence renders early-stage breast cancers (i.e., tumors that have not spread beyond breast and nearby lymph nodes) amenable to endocrine therapy as the key therapeutic approach2. Valid adjuvant options include the selective estrogen receptor modulator tamoxifen that competes with estrogen for the binding to ERα to block signaling, and the 3rd generation aromatase inhibitors (AIs) anastrozole, letrozole, and exemestane that remove estrogen from the signaling cascade by blocking its synthesis3. AIs evolved as the standard-of-care for postmenopausal women4 and have become an option for premenopausal women together with concurrent ovarian suppression5. Their mechanism of action in lowering estrogen levels is particularly important for postmenopausal women, as AIs target the aromatase enzyme (CYP19A1) in peripheral nonglandular tissues (and breast tumor) to block the conversion of testosterone and androstenedione to 17β-estradiol and estrone, respectively3. Current guidelines recommend 5 to 10 years of treatment to maximize risk-reduction2. During this period and beyond, women continue to be at risk of recurrence and death that extends for decades following treatment6. Therefore, additional biomarkers for the proper selection of drug treatment regimens are a high research priority.
Inherited germline variants account for the inter-patient variability of pharmacokinetic and pharmacodynamic drug effects. In the case of AIs, putative predictors include polymorphisms at drug metabolizing enzymes such as CYP3A4 and UGT1A4 for anastrozole, CYP2A6 for letrozole, as well as CYP3A4 and UGT2B17 for exemestane, and associations have been reported with regards to systemic AI concentrations, systemic estrogen concentrations, treatment efficacy, and toxicities7. However, despite compelling biological and pharmacological rationales, the evidence of linking the magnitude of estrogen suppression and AI efficacy with the patients’ constitutional background remains a challenge. For example, comparative analyses of patients with anastrozole and exemestane (MA.27 trial) as well as letrozole treatment (PreFace trial) linked insufficient estrogen suppression with a 2.2-fold increased risk of early breast cancer events (p-value = 5E-04)8. A corresponding pharmacogenomic Genome-Wide Association Study (GWAS) identified a common variant (rs6981827) in the human CUB And Sushi multiple domains 1 gene (CSMD1, alias PPP1R24)9 to be associated with the changes in estrogen levels under anastrozole, and a weakly linked expression Quantitative Trait Locus (eQTL) single-nucleotide polymorphism (SNP rs6990851) that was associated with breast cancer-free interval in anastrozole- and exemestane-treated patients. Moreover, the risk of breast cancer recurrence in patients treated with anastrozole and exemestane has been linked to SNPs in the long non-coding RNA MIR2052HG10. Both findings were underpinned by mechanistic studies in that the CSMD1 genotype correlated with CYP19A1 expression in an anastrozole-dependent fashion through transcription regulation9. In the case of MIR2052HG, variant genotypes exhibited increased ERα binding to estrogen response elements relative to wild-type genotypes, thereby downregulating ERα transcription in vitro, which may affect the risk of breast cancer recurrence in women treated with AI10.
Despite important insights, the identification of relevant AI outcome predictors remains a challenge given the scarcity of suitable study and replication cohorts for early breast cancer. Association studies in the breast cancer endocrine treatment setting are particularly challenging, as the lengthy 5-year treatment and further 5 to 10 year follow-up period during which recurrence or death events may occur, limit the availability of such cohorts. In the current work we performed a two-stage approach, utilizing the database of the Breast Cancer Association Consortium (BCAC) with its comprehensive clinical, epidemiologic, and genetic data set11,12,13 for discovery, and independent clinical cohorts (MA.27, PreFace, KARMA/pKARMA, MCBCS, IKP211) for validation analysis to explore potential associations between common germline variants and survival endpoints in AI-treated patients. The aim of our study was to identify putative pharmacogenetic/-genomic polymorphisms in genes, that based on their suspected role in endocrine resistance and converging/interconnected mechanisms may contribute to AI treatment outcome in early-stage ER-positive breast cancer.
Results
Candidate gene association (CGA) study in AI-treated patients (discovery analysis – stage 1)
Candidate gene association analysis was performed in 2789 AI-treated breast cancer patients selected from the BCAC database (version 12) (Fig. 1). Details on patients, their clinical data and tumor characteristics are given in Table 1. Median follow-up time was 7.2 years, and event rates within a 15-year follow-up period were 14.4% (overall survival, OS), 15.2% (distant relapse-free survival, DRFS), and 16.8% (relapse-free survival, RFS). Analyses were performed using genotype data of 753 selected genes (Supplementary Data 1) related to endocrine resistance and converging interconnected mechanisms. Subgroup analyses included 1335 patients treated with anastrozole (47.9%) and 689 patients treated with letrozole (24.7%). No subgroup analysis was performed for patients treated with exemestane due to low case numbers (Fig. 1).

The BCAC database (version 12) comprised 92,933 patients with a confirmed diagnosis of invasive ER-positive breast cancer who are of European descent (based on genotype data) and older than or equal to 18 years of age. Of these, 27,887 patients had early-stage disease with documented adjuvant endocrine therapy (AI, Tamoxifen, switch therapy, or unspecified) and complete follow-up data. The subgroup of AI-treated patients analysed in this study comprised 2789 women of which 1335 had received anastrozole, 689 letrozole, and 114 exemestane. For 570 patients, AI treatment was not further specified and 81 patients had received an additional endocrine treatment. AI Aromatase Inhibitor, OS Overall survival, DRFS Distant relapse free survival, RFS Relapse free survival.
Primary endpoints OS, DRFS, and RFS
All AI-treated patients: Unadjusted analysis revealed 47, 65, and 54 variants with an association p-value ≤ 1E-04 for OS, DRFS, and RFS, respectively (Supplementary Data 2; minor-allele frequency (MAF) ≥ 5%; 125 non-overlapping variants in total). Their corresponding chromosomal regions are highlighted in the Manhattan plots of Fig. 2a and include the following gene regions: For OS, top associated variants mapped to the CELA2B/CASP9 (n = 16 variants) and NR1I2/GSK3B loci (n = 30). For DRFS, variants in the CELA2B/CASP9 region were also among the top hits (n = 52), including 37 variants that annotate to the intronic and 5’ region of CASP9. Other DRFS top hits included variants in LRP1B (n = 8) and the CARMN/MIR143HG locus (n = 2). For RFS, top hits comprised variants at CELA2B/CASP9 (n = 16; all among DRFS top hits), LRP1B (n = 31; including the 8 variants associated with DRFS), and CARMN/MIR143HG (n = 2) gene regions. Similar results were observed for the top associated variants in analyses adjusted for patient characteristics (age, menopausal status, and family history of breast cancer in a first degree relative), tumor characteristics (size, grade, nodal status, stage, and progesterone receptor status), treatment (AI, adjuvant chemotherapy, trastuzumab, and radiation), and surgery type (Supplementary Data 3).
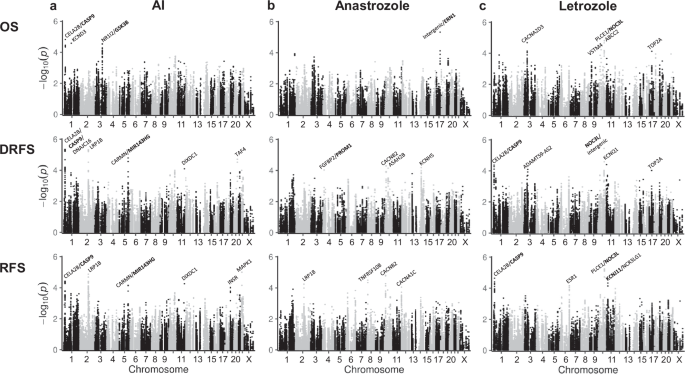
The Manhattan plots display the results of unadjusted analyses (stratified by country and corrected for genetic principal components). The y-axis shows the –log10 meta-analysis p-values and the x-axis the chromosomal positions of the variants. Variants are located within 753 selected candidate genes or surrounding gene region (defined as +/- 10 kb from gene start/end). For variants with association p-value ≤ 1E-04, the corresponding gene symbols are displayed on top (“intergenic” in case no gene was annotated). Genes marked in bold were part of the 753 candidate genes. Analysis of (a) patients treated with any AI (n = 2789) and approximately 170,000 variants with MAF ≥ 0.05, (b) patients treated with anastrozole (n = 1335) and approximately 138,000 variants with MAF ≥ 0.1, and (c) patients treated with letrozole (n = 689) and approximately 138,000 variants with MAF ≥ 0.1. Survival endpoints include OS (overall survival), DRFS (distant relapse-free survival), and RFS (relapse-free survival).
For the top hits, we scrutinized eQTLs to support the selection of candidate variants to be further pursued in validation analysis. No survival eQTLs were revealed in PancanQTL analysis, which however was not focused on AI-treated patients. Yet, PancanQTL and FUMA (Functional Mapping and Annotation) tools identified the top variants of the CELA2B/CASP9 region as cis-eQTLs associated with CASP9 expression in breast cancer, breast mammary tissue as well as numerous other normal tissues and cancer entities. GSK3B top hits were revealed as cis-eQTLs in breast mammary tissue, non-breast tumor entities, and various normal tissues. Furthermore, cis-eQTLs were identified for LRP1B and CARMN/MIR143HG in non-breast tissues. We then used KM Plotter to investigate whether expression levels of genes identified in the eQTL databases were correlated with OS in ER-positive, endocrine treated breast cancer patients. Here, high CASP9, GSK3B, and LRP1B expression levels were associated with better survival. For GSK3B, an opposite effect was observed when patients with chemotherapy were excluded. Moreover, stratification by molecular subtype demonstrated that high GSK3B expression was associated with a better outcome in Luminal B but worse outcome in Luminal A patients (Supplementary Fig. 1-3 in the Supplementary Information).
Anastrozole-treated patients: We identified n = 2 (OS), n = 4 (DRFS), and n = 8 (RFS) top associated variants in unadjusted analysis (Supplementary Data 4; p-value ≤ 1E-04 and MAF ≥ 10%). Their corresponding chromosomal regions are indicated in Fig. 2b: For OS, top associated variants were located in an intergenic region 5’ of the ERN1 gene (n = 2). DRFS top associated variants included singletons at ASAH2B, KCNH5, CACNB2, and in the FGFBP2/PROM1 locus. Associated variants for RFS mapped to CACNB2 (n = 4) as well as to TNFRSF10B (n = 2), CACNA1C, and LRP1B (both singletons). Note that the single LRP1B hit (rs77397926; MAF ~ 40%) is not linked to the LRP1B variants identified in the all AI analysis.
Letrozole-treated patients: Unadjusted analysis showed 22, 9, and 33 top associated variants for OS, DRFS, and RFS, respectively (Supplementary Data 5; p-value ≤ 1E-04 and MAF ≥ 10%). Their corresponding chromosomal regions are highlighted in Fig. 2c: For OS, top associated variants mapped to the CACNA2D3 (n = 4), ABCC2 (n = 4), and PLCE1/NOC3L (n = 12) loci. Linked top variants in the latter gene region were also identified in DRFS (n = 2) and RFS (n = 22) analyses, however they showed rather low MAFs (iCOGS: ≤ 8.5%, OncoArray: ≤ 11.4%). In addition, top associated variants in DRFS and RFS analyses were identified at the CELA2B/CASP9 locus (n = 3 and 5, respectively), all of which were also among the DRFS top hits in the all AI analyses. Further RFS associated top variants were located at the ESR1 (n = 4) and KCNJ11/NCR3LG1 (n = 2) loci.
Secondary endpoints breast cancer-specific survival (BCSS), distant recurrence-free interval (DRFI), and recurrence-free interval (RFI)
Unadjusted analyses of all AI-treated patients for the secondary endpoints BCSS, DRFI, and RFI revealed 14, 3, and 3 variants with association p-value ≤ 1E-04, respectively (Supplementary Fig. 4 in the Supplementary Information, Supplementary Data 6; MAF ≥ 5%). Of the 14 BCSS top hits, 13 were tightly linked variants in CACNA1D and the remainder was located in the ERN1 gene region. Top variants in DRFI analysis comprised 3 variants with association p-value ≤ 1E-04 (2 variants in the FGFBP2/PROM1 gene region and a singleton in the KCNK9 gene), and another n = 85 with p-value ≤ 5E-04. These included further 9 variants in the FGFBP2/PROM1 gene region, top hits in the CASP9/CELA2B locus (n = 17) and LRP1B (n = 7) as well as variants in ESRGG (n = 6), NEDD9 (n = 7), and VCAN (n = 3). Top RFI hits comprised 67 variants with p-value ≤ 5E-04, including 16 variants in LRP1B and 11 variants in VCAN (including one SNP with p-value ≤ 1E-04), as well as singletons each in DIXDC1 and KCNK9 with p-value ≤ 1E-04.
Selection of candidate variants for validation
The primary objective of our study was the validation of selected top variants identified in the OS, DRFS, and RFS discovery analyses of all AI-treated patients, using independent AI treatment cohorts. Candidate selection was based on a p-value-based filtering strategy (association p-value ≤ 1E-04) and a power/sample size estimation approach that accounts for “winner’s curse bias”. In total, six SNPs were selected for primary analysis, including rs3820071 (DRFS, RFS) and rs6685648 (DRFS) in the CELA2B/CASP9 locus, rs10496860 (DRFS, RFS) in LRP1B, rs3107669 (OS) in GSK3B, as well as rs353298 (DRFS, RFS) and rs353296 (RFS) in the CARMN/MIR143HG gene region (Table 2). The investigation of the candidates for the respective remaining survival endpoints as well as in the anastrozole and letrozole treatment subgroups was part of our secondary objectives. Further secondary objectives included the examination of top hits additionally identified in the anastrozole and letrozole treatment subgroup analyses as well as in the analyses of the secondary endpoints BCSS, DRFI, and RFI. For the former, we selected rs11012983 (CACNB2; anastrozole-treated patients) and rs827423 (ESR1; letrozole-treated patients; Supplementary Table 1a in the Supplementary Information) applying similar filter criteria as for the primary analyses. Selected BCSS, DRFI, and RFI candidates covered variants in CACNA1D (rs2276836), ESRRG (rs830321), NEDD9 (rs4713432), and VCAN (rs251124 and rs2541311), where we relaxed the p-value criterion to p ≤ 5E-04 (Supplementary Table 1b in the Supplementary Information).
Validation analysis of selected candidates (stage 2)
The five validation cohorts comprised 8557 AI-treated patients (2673 patients treated with anastrozole, 2973 patients with letrozole, 2253 patients with exemestane, and 658 patients with unspecified AI treatment or switch regimens). Details on clinical and histopathological characteristics as well as treatment regimens are given in Table 1. Median follow-up times for MA.27, PreFace, and IKP211 were 4.1, 5.3, and 5.5 years, and exceed 10 years for KARMA/pKARMA and MCBS, respectively. Validation analyses of the six primary candidates were performed similar to the discovery analysis. Concerning our primary objective, we observed effect sizes (per-allele HR) up to 1.2; however, we did not identify any significant associations in the analysis of all AI-treated patients (Table 3, Fig. 3a–c, Supplementary Fig. 5 in the Supplementary Information). Lowest p-values were observed for the CARMN variants rs353298 (DRFS and RFS, p-value = 0.280 and 0.108) and rs353296 (RFS, p = 0.084), however with opposite effect directions compared to the discovery analysis. This trend was consistent for three cohorts (PreFace, MCBCS, and IKP211), but not for MA.27. Particularly for rs353296, effects were heterogeneous across the validation cohorts (I2 > 52%, heterogeneity p-value < 0.1; Fig. 3c). Considerable study heterogeneity was also observed for GSK3B variant rs3107669 (I2 = 49.7%, heterogeneity p-value = 0.078; Fig. 3a, upper part). A detailed examination of rs3107669 revealed that the exclusion of one cohort, i.e., MA.27, considerably reduced heterogeneity, and the corresponding hazard ratio (HR) was shifted towards the discovery finding (HR = 1.17, 95% confidence interval (CI) = [1.00, 1.37], meta-analysis p-value = 0.045, I2 = 0%, heterogeneity p-value = 0.321; Fig. 3a, lower part). All other variants showed homogeneous effects in the analysis of all AI-treated patients (I2 < 25%, heterogeneity p-value > 0.2; Supplementary Fig. 5 in the Supplementary Information). Secondary adjusted analyses as well as sensitivity analyses did not reveal substantially different results (Supplementary Table 2 in the Supplementary Information).

Forest plots depicting per-allele hazard ratios (x-axis) and corresponding confidence intervals for (a) rs3107669, (b) rs353298, and (c) rs353296 in eligible validation cohorts. The size of the squares reflects the study size. The KARMA/pKARMA studies were omitted in DRFS and RFS analyses due to delayed study entry of nearly all 643 patients ( > 0.5 years after diagnosis). Diamonds represent results of random effects (RE) meta-analyses in validation cohorts and of the discovery analyses in BCAC patients, respectively. Endpoints are OS (overall survival), DRFS (distant relapse-free survival), and RFS (relapse-free survival).
Similar results for the six primary candidates were also observed in treatment subgroups analyses. No significant associations were obtained in the unadjusted analysis of anastrozole-treated patients (n = 2673; MA.27, MCBCS, IKP211; Supplementary Fig. 6 in the Supplementary Information). The corresponding letrozole analysis (n = 2973; PreFace, MCBCS, IKP211) revealed a low association p-value for the CARMN variant rs353296 with RFS and no study heterogeneity, but again an opposite effect direction was observed when compared to the discovery finding (HR [95% CI] = 0.69 [0.54, 0.89], meta-analysis p-value = 0.003, I2 = 0%, heterogeneity p-value = 0.452). Opposite effect directions were also observed for the other endpoints (OS, DRFS) and for rs353298. The effect direction for GSK3B variant rs3107669 in the letrozole analysis matched that of the discovery finding, however the effect was insignificant and heterogeneous in the validation cohorts (OS: HR [95% CI] = 1.42 [0.79, 2.56], meta-analysis p-value = 0.237, I2 = 65.3%, heterogeneity p-value = 0.056; Supplementary Fig. 7 in the Supplementary Information). Further secondary objectives in our validation analysis were concerned with the examination of top hits identified in the treatment subgroup analyses as well as in the analysis for the additional survival endpoints BCSS, DRFI, and RFI considering breast-cancer specific death. The CACNB2 variant rs11012983 did not show a significant effect in the anastrozole analysis (all p-values > 0.1, Supplementary Fig. 6 in the Supplementary Information). In the letrozole analysis, the ESR1-SNP rs827423 showed a similar trend in discovery and validation cohorts; however this trend was not significant (RFS: HR [95% CI] = 0.87 [0.70, 1.08], meta-analysis p-value = 0.208, I2 = 3.9%, heterogeneity p-value = 0.402; Supplementary Fig. 7 in the Supplementary Information). Examination of the five BCSS, DRFI, and RFI candidates also did not reveal any significant results in the validation studies (all p-values > 0.1) (Supplementary Fig. 8 in the Supplementary Information).
Discussion
This study addressed the identification of potential pharmacogenomic predictors for AI treatment outcome in patients with hormone receptor-positive early breast cancer. To do so, we leveraged one of the largest clinical, epidemiologic, and genetic breast cancer data sets available from BCAC to perform hypothesis-driven pharmacogenomic analyses, using OS, DRFS, and RFS as the main survival endpoints. Our two-stage candidate gene approach focused on patients that met the clinical definition for adjuvant AI treatment as reflected in the favorable event rates in both discovery and validation cohorts (Table 1). In this study, we analyzed approximately 170,000 genetic variants located in 753 gene regions that have been selected based on their proposed roles in breast cancer endocrine resistance and/or related and intersecting pathways, in which ER constitutes the principal signal transduction pathway14,15,16,17,18.
Results from the discovery analysis of 2789 AI-treated patients highlighted 125 top associated loci for the main endpoints OS, DRFS, and RFS (p-value ≤ 1E-04) including variants located in the regions of CASP9 (Chr. 1), LRP1B (Chr. 2), GSK3B (Chr. 3), and CARMN (Chr. 5), but not in any known pharmacogenes. Published candidates from survival GWAS or CGA studies of ER-positive, early-stage, AI-treated breast cancer patients were not significant in our analysis9,10,19,20. Rather, OS associations were particularly evident for CASP9 and GSK3B, with multiple closely linked variants (r2 > 0.9), respectively. For DRFS and RFS, multiple associations were again observed for CASP9, but also for LRP1B (Fig. 2, Supplementary Data 2). Supportive evidence for the top variants’ potential modulatory role in breast cancer outcome came from queries on eQTL in tumor and normal tissue. For CASP9, encoding a tumor suppressor caspase and recognized as a hub gene in inflammatory breast cancer21, the variants were associated with its mRNA expression. High CASP9 expression in turn showed a favorable role for OS in patients with ER-positive breast tumors based on RNA-seq data22, which is in line with results of a gene-chip-based analysis21. This remained statistically significant when the analysis was limited to patients with a good prognosis (i.e., without chemotherapy; Supplementary Fig. 1 in the Supplementary Information). Similar associations have been observed for the neighboring genes CELA2B and DNAJC16, highlighting a putative role of this locus in the survival of early-stage breast cancer patients.
Likewise, for GSK3B, a unique serin/threonine kinase that acts as a signaling node at the intersection of multiple cancer-related pathways such as NFκB, Wnt/ß-catenin, MEK/ERK, PI3K/Akt and Notch23, top variants were identified as cis-eQTLs that impact on gene expression in mammary tissue. While GSK3B is subject to bidirectional regulation contingent on phosphorylation at Y216 (activation) and S9 (inhibition), a dual role as tumor suppressor and promoter has been noted24. The tumor-suppressive role is mainly attributed to its activated state resulting in the downregulation of proto-oncogenic genes (e.g., MYC, CCND1) mediated by the blockage of ß-catenin signaling via its phosphorylation and subsequent proteasomal degradation and/or direct regulation at protein level (e.g., Cyclin D1)24. Notably, Cyclin D1 is among the most commonly overexpressed proteins in breast cancer and a combinatory role of Cyclin D1 and GSK3B expression in survival has been suggested with high GSK3B levels increasing the risk for distant relapse and low levels being more favorable25. With regard to gene expression, we observed that high GSK3B levels showed a protective effect on the OS of patients with ER-positive breast tumors, while an inverse effect was revealed in the subgroup of patients with Luminal A (but not Luminal B) tumors, stressing GSK3Bs’ putative dual role in the biology of these distinct molecular tumor subtypes.
Although our discovery analysis and in silico resources support a putative role of CASP9 and GSK3B in breast cancer and AI treatment outcome, none of the six primary candidate variants have been validated in the independent breast cancer AI treatment cohorts. A reason for the failed replication could be that these are false positives of our discovery analysis. On the other hand, we may have missed polymorphisms with low frequency or weaker effects on the survival endpoints. Yet, the objective of the first stage of the analysis was the identification of a candidate set that would most likely include the top true survival-associated polymorphisms, which would then be analyzed in independent validation cohorts with sufficient power to correct for multiple testing and winner’s curse.
While the strength of our study is its two-stage design that meets the gold standard of genetic association studies26, and its large size of more than 11,300 AI-treated breast cancer patients, it is at the same time subject to several limitations intrinsic to large drug treatment outcome related studies27,28. These comprise differences in study design or clinical characteristics such as age at diagnosis, histopathological parameters, treatment regimens, as well as length of follow-up, that all may contribute to heterogeneity among study cohorts. Moreover, studies could potentially suffer from underreporting of events, and commonly investigated endpoints may be limited surrogates of endocrine treatment efficacy in pharmacogenomic investigations. In terms of heterogeneity, we noticed considerable differences for clinical characteristics and treatment regimens across the validation cohorts. While adjusted analyses did not reveal substantial differences compared to unadjusted analysis, we observed especially for GSK3B rs3107669 that the exclusion of MA.27 considerably reduced heterogeneity, with the corresponding HR shifting in the direction of our discovery results (OS: HR [95% CI] = 1.17 [1.00, 1.37], p-value = 0.045, I2 = 0%, heterogeneity p-value = 0.321; Fig. 3a). Thus, we cannot rule out that heterogeneity across the study cohorts has hampered, at least to some extent, our validation effort. In this context, it should be noted that genetic variants with similar effects across different AI treatment regimens have been described in the literature, but even opposite effect directions were reported. For example, MIR2052HG variants were associated with breast cancer-free interval (BCFI) in the combined anastrozole and exemestane treatment group of the MA.27 trial10, however in anastrozole- and exemestane-only analyses opposite BCFI effects of multiple other genetic variants were identified29. Yet, the previously described variants assessed in our discovery analyses were not strongly associated, neither in all AI-treated patients nor in anastrozole-only treated patients (all unadjusted p-values > 0.04). This may be due to the use of other endpoints (OS, DRFS, RFS) and the limited sample size of our discovery cohort, particularly with regard to anastrozole-only (n = 1335) and letrozole-only (n = 689) analyses. Of note, our subgroup analyses revealed only few additional candidates as compared to the all-AI analysis (n = 2789), a reason why we chose the latter as primary analysis.
It is important to view our findings within the context of reported challenges and uncertainties inherent to treatment outcome association studies that may limit their value27,28. We support the view that better-quality cohort appropriateness is key for the detection of treatment-related effects and appreciate that efficacy (survival) endpoints such as OS, DFS, or RFS differ from phenotype endpoints (e.g. disease risk), as they are liable to additional confounders related to patients’ drug treatment history (e.g. type, duration, adherence, etc). Thus, it will be advisable to control for these in the future to warrant detectability of small treatment-related effects.
A particular concern may relate to the specific drug action inherent to endocrine treatment, that currently is not well-captured by standard descriptive variables and endpoints. In contrast to cancer treatments that act via cytotoxic effects28, the efficacy of endocrine treatment including AI is subject to a cytostatic effect that persists over time. At a glance, a minimum of 5-year treatment is required to keep micrometastases in the women’s body at check, which is conveyed by a carry-over effect beyond the actual drug intake until resistance occurs4,6,16. Clinical events such as relapse and death follow an evolving micrometastatic phenotype via selection and expansion of a resistant subpopulation of tumor cells and reprogramming of the tumor cells’ biological pathways. Accordingly, the tumor cell itself potentially holds promise for a more precise efficacy endpoint than survival. Despite laboratory proof of concept of an adapting molecular landscape during AI treatment14,15,16,17,18, and clinical proof of concept that this adaptive process is a critical determinant of endocrine treatment outcomes30, the notion of an application of the dynamic tumor molecular landscape as a measurable AI-related endpoint may be premature. Yet, innovation on micrometastasis detection and manipulation are on the rise31,32 and it may therefore be reasonable to speculate that proper techniques for the assessment of quantifiable endpoints may become available.
Although we did not meet our primary objective, our work generated important findings from which we conclude that efforts towards further standardization of endocrine treatment pharmacogenomic studies for ER-positive early-stage breast cancer need to be intensified. A lesson learned is to widen the view towards innovative approaches to better capture treatment efficacy. The latter requires revisiting the way we define informative endpoints, which can only be achieved via controlled longitudinal clinical trials with sufficiently long follow-up assessing drug-related parameters including adherence that would then be complemented by the monitoring of measurable molecular tumor cell parameters.
Methods
Breast Cancer Patients
Patient characteristics, clinical data (e.g., treatment, follow-up, and survival endpoints), and genotype data have been retrieved from established data repositories including hospital-based and case-control studies, as well as prospective clinical trials and observational studies for the investigation of an association between common germline variants and patients’ outcome upon AI treatment. Study subjects include patients that had received an AI, i.e. anastrozole, letrozole, or exemestane for the treatment of their primary invasive early breast cancer that was ERα and/or progesterone receptor (PR) positive. Patient origin, size of discovery cohort (stage 1) and validation cohorts (stage 2), as well as extraction procedures are briefly summarized as follows with more details provided in Supplementary Data 7 and 8, respectively.
Discovery cohort (stage 1)
We used data from selected breast cancer patients from studies participating in the Breast Cancer Association Consortium (BCAC) stored in the BCAC database at the University of Cambridge, UK. Data have been retrieved from database version 12 (07/2019) that in addition to clinical, epidemiological, and genotype data includes information about histopathology, survival, and treatment pooled and harmonized at the Netherlands Cancer Institute prior to integration12,13. Participating studies were approved by respective ethics committees, and patients’ informed consent was obtained from all patients (Supplementary Data 7). Female breast cancer patients have been first selected based on a diagnosis of primary invasive breast cancer, European ancestry (based on genotype data), age at diagnosis ≥ 18 years, and ER-positive tumor status (n = 92,933; Fig. 1). To match the study sample to the definition of an intended 5-year adjuvant endocrine treatment setting, patients not having received endocrine treatment or with unknown treatment status, metastatic disease at diagnosis or unknown metastatic status, as well as unknown or incomplete follow-up information on survival endpoints have been subsequently excluded (n = 65,046 patients; Fig. 1). The remaining 27,887 patients (30%) included 2789 AI-treated patients that served as the discovery cohort in this current investigation. Figure 1 illustrates the selection process, and Supplementary Data 7 provides details of the 25 contributing BCAC studies from Europe, USA, and Australia. Among the AI-treated patients, the majority had received anastrozole (n = 1335; 47.9%) or letrozole (n = 689; 24.7%) in line with the then in force standard of care in their countries of origin, with fewer patients having received exemestane (n = 114; 4.1%). For a large subgroup, AI treatment was not further specified (n = 570; 20.4%) or patients in addition to AI had received additional endocrine treatment (n = 81; 2.9%). The median follow-up of the 2789 AI-treated patients was 7.2 years (Interquartile Range (IQR): 3.9 to 10.5 years, Table 1). Note, patients treated with tamoxifen (Fig. 1) are subject to separate analyses and are not included in this current work.
Validation cohorts (stage 2)
Contributing cohorts were selected based on availability and study design to match the aforementioned criteria for adjuvant AI endocrine treatment. The total cohort comprised 8557 AI-treated patients from five independent studies including AI outcome-related prospective randomized and observational clinical trials (MA.2733, PreFace34, IKP21135,36) as well as case-control and cohort studies (KARMA/pKARMA37, MCBCS38). Overall, there were 2673 patients treated with anastrozole (MA.27, MCBCS, IKP211), 2973 patients treated with letrozole (PreFace, MCBCS, IKP211), and 2253 patients treated with exemestane (MA.27, MCBCS, IKP211), as well as 658 patients with unspecified AI treatment or switch regimens. Median follow-up times for MA.27, PreFace, and IKP211 were 4.1, 5.3, and 5.5 years, and exceeded 10 years for KARMA/pKARMA and MCBS, respectively (Table 1). Details of each individual study regarding study design, recruitment, type of AI treatment, and follow-up are given below and in Supplementary Data 8.
MA.27: The MA.27 trial (ClinicalTrials.gov identifier NCT00066573) is a phase III cooperative group study, designed as multicenter, multinational, randomized open-label trial. The trial included postmenopausal women with histologically confirmed and completely resected stage I-III breast cancer (AJCC Version 6) that was ERα and/or PR positive. Patients were randomized to 5 years of anastrozole or exemestane for the testing of superior efficacy of the latter with regard to event-free survival33. Following approval by health regulatory authorities and the centers’ regulatory approval boards, the study enrolled 7575 women, among whom North American patients provided a blood sample and gave consent for genetic testing. Research was performed after approval by local institutional review boards in accordance with assurances filed with, and approved by the Department of Health and Human Services10. Our validation study used baseline and clinical as well as relevant genetic data of 4408 patients, of whom 2195 patients had received anastrozole and 2213 patients exemestane. Median follow-up time was 4.1 years (IQR: 3.9-5.0 years; Table 1).
PreFace: The PreFace study (ClinicalTrials.gov identifier NCT01908556) is a single-armed, multicenter, open-label, phase IV trial for the evaluation of predictive factors of the effectivity of AI therapy. The study recruited 3483 postmenopausal early-stage breast cancer patients with a hormone receptor-positive breast cancer (without distant metastases) across 220 study sites in Germany. Patients were assigned to treatment with 2.5 mg daily letrozole after the completion of standard therapies (surgery, chemotherapy, and radiotherapy) in accordance with national guidelines. Treatment was continued until disease recurrence or up to a maximum of 5 years34. All patients provided blood samples for genetic testing of putative associations between polymorphic loci and survival as well as quality of life. The study was approved by Ethics Committee of the Medical Faculty of the University of Erlangen-Nuremberg (Number 25_2008) and all other relevant ethics committees. Our validation study included baseline, clinical, and relevant genetic data of 2761 letrozole-treated patients. Median follow-up time was 5.3 years (IQR: 4.7-5.7 years; Table 1). Patient characteristics selected for this current study did not differ from the patient characteristics of the main study34.
KARMA/pKARMA: The Karolinska Mammography project (KARMA) is a prospective cohort study of women attending mammography screening or clinical mammography at four hospitals in Sweden37 for the assessment of epidemiological and molecular breast cancer risks. Between 10/2010 and 03/2013, 70,877 women were enrolled, including 3000 incident breast cancer cases diagnosed at cohort entry. All study participants gave informed consent and ethics approval was obtained from the ethical board at the Karolinska Institute. Approximately 53% of the recruited women were postmenopausal. The cohort is matched to relevant Swedish medical quality registries for biannual follow-up. There is an estimated annual rate of 250 new breast cancer diagnoses. Follow-ups recorded at the national registries include information on the use of medication, secondary diagnoses, and death events. Similarly, pKARMA is a case-control study with incident breast cancer cases recruited between 01/2001 and 12/2008 in the Stockholm/Gotland area by the Stockholm Breast Cancer Registry. Recording of survival data follows that of the KARMA study. Our validation study included altogether 643 AI treated patients from both cohorts with a median follow-up time of 10.8 years (IQR: 7.8-12.2 years). Specific information on the type of AI treatment was not available (Table 1).
MCBCS: Incident breast cancer cases from 6 US states (MN, WI, IA, IL, ND, SD) were recruited within the framework of a hospital-based case-control study for questionnaire-based, molecular risk factor analyses at the Division of Medical Oncology at the Mayo Clinic in Rochester, MN (02/2001-06/2005)38. Cases were diagnosed within the previous six months and no prior history of cancer (except non-melanoma skin cancer), and clinical and treatment data as well as follow-up and survival data were retrieved by medical record abstraction during annual follow-up surveys. Our validation study used data from 392 patients that were treated with an AI. Of these, 335 received anastrozole, 38 letrozole, and 19 exemestane. Median follow-up time was 10.1 years (IQR: 7.0-11.7 years; Table 1).
IKP 211 Study: The IKP211 study (German clinical trials registry identifier DRKS00000605) is a prospective multi-center observational breast cancer study for the investigation of endocrine treatment efficacy and toxicity. The study recruited 1286 postmenopausal patients with a diagnosis of early-stage invasive breast cancer that was ER and/or PR positive. Patients received 5 years of adjuvant treatment, either with tamoxifen or an AI or switch from tamoxifen to AI or vice versa in line with local clinical practice and national guidelines. Patients were recruited at 37 breast centers and hospitals. Most patients were followed up during a 10 year study extension period for the documentation of recurrence and survival. All patients provided blood and plasma samples for genetic testing and drug metabolite measurements35,36. Ethics approvals were obtained from the Ethics Committee of the University of Tübingen, as well as respective local committees of the participating centers. Follow-up was recorded for 1159 patients, of whom 550 patients were treated with an AI (48%) and the remainder with tamoxifen (292 patients; 19%) or switch therapy (317 patients; 27%). AI treated patients in most cases received letrozole (n = 418). Our validation study used clinical and relevant genetic data of 353 AI treated patients that were composed of 143 anastrozole-, 174 letrozole-, and 21 exemestane-treated patients, as well as 15 patients with AI-to-AI switch therapy. Note, 210 letrozole-treated patients were not considered in this current validation due to an overlap with the PreFace study that was conducted in parallel. Median follow-up time was 5.5 years (IQR: 4.6-10.1 years; Table 1).
Ethics statement
The study was performed in accordance with the Declaration of Helsinki. All individual studies included in the analyses were approved by the appropriate institutional ethical review boards following their national guidelines for informed consent; the details are provided in Supplementary Data 7 and 8.
Genotyping, quality control, and imputation of genetic variants
Discovery cohort
Genotypes were established as part of the Collaborative Oncological Gene-Environment Study (COGS) and the OncoArray project using the Illumina iSelect array (211,155 genetic variants) and the Illumina OncoArray-500K BeadChip (533,631 genetic variants), respectively11,39. The complete list of genotyped SNPs located in the 753 candidate gene regions is provided in Supplementary Data 1. Non-genotyped variants were imputed based on the 1000 Genomes Project Phase 3 release (October 2014) as reference panel. More details on genotyping and imputation of genetic variants as well as quality control are described elsewhere12,39,40. Our main discovery analysis in 2789 patients considered approximately 170,000 variants in 753 candidate gene regions that had a MAF ≥ 5% and were typed or imputed with imputation r2 ≥ 0.8 in at least one of the datasets (iCOGS and OncoArray). Due to lower sample sizes, the anastrozole (n = 1335) and letrozole (n = 689) subgroup analyses have been restricted to variants with MAF ≥ 10%, resulting in approximately 138,000 variants.
Validation cohorts
MA.27 samples
Genotyping was performed on three different platforms: Illumina Human610 Quad BeadChip, Illumina Human OmniExpress, and Illumina Human OmniExpressExome. Imputation was performed across the three platforms separately. Details on genotyping and imputation of genetic variants as well as quality control are described in Ingle et al.10 and literature cited therein. PreFace samples: Genotyping was also established as part of the OncoArray project using the Illumina OncoArray-500K BeadChip and the same imputation methods as described above for the discovery cohort. KARMA/pKARMA and MCBCS samples: Genotyping data (including typed and imputed variants) were retrieved from BCAC database version 12. IKP211 samples: Genotyping was performed on the Illumina Infinium OmniExpress 12/24 BeadChips platform (Illumina, Singapore). Quality control and imputation were performed similar to Khor et al.41. In brief, samples with call rates < 95% or showing extremes of heterozygosity as well as variants with call rates < 90%, MAF < 1%, or Hardy Weinberg Equilibrium p-value < 1E-06 and non-autosomal markers were excluded. The McCarthy Group imputation preparation tool (https://www.well.ox.ac.uk/~wrayner/tools/index.html, version 4.2.11) was applied for further quality control and filtering, and SHAPEITv242 as well as IMPUTE v2.3.243 were used for pre-phasing and genotype imputation, respectively, using the 1000 Genomes Phase 3 reference panel.
Selection of candidate genes
Candidate genes have been selected based on their putative roles in distinct biological pathways of endocrine resistance and converging/intersecting pathways. They include on the one hand genes associated with disease progression and breast cancer relapse during adjuvant endocrine treatment and thereafter. On the other hand, based on in vitro models, they include genes involved in the dynamic reconfiguration of the tumor cells’ survival program during long-term estrogen deprivation, with ER constituting the principal signal transduction pathway for replication and death14,15,16,17,18. This knowledge framework served as a source for the selection of 753 genes investigated in this current CGA study (Supplementary Data 1). In brief, selected genes operate within the following context: endocrine resistance (n = 217), microRNAs of endocrine resistance, their targets and biosynthesis (n = 262), cancer-related channels, particularly potassium channels in endocrine resistance (n = 144), cholesterol biosynthesis and transport (sterol pathway; n = 52), CYP3A4 drug metabolism and ER signaling (n = 53), as well as other cell adhesion, surface, and transmembrane proteins including receptors and stem cell markers that may contribute to endocrine resistance (n = 25). Variants have been retrieved from 10 kb upstream to 10 kb downstream of the chromosomal positions of the 753 candidate genes. Detailed information is given in Supplementary Data 1.
Survival endpoints
Outcome measures followed the standardized definitions for efficacy endpoints in adjuvant breast cancer trials as described by the STEEP system44. They are defined as follows, with deviations from STEEP indicated in italics. Time-to-event was calculated from the date of diagnosis for all cohorts except the MA.27 trial, for which the date of randomization was used instead33.
Primary endpoints
Overall survival (OS)
Time from diagnosis/randomization to death of any cause (death from breast cancer, death from nonbreast cancer cause, or death from unknown cause). Distant relapse-free survival (DRFS): Time from diagnosis/randomization to time of first documented distant recurrence or death of any cause. Relapse-free survival (RFS): Time from diagnosis/randomization to first documented recurrence (invasive ipsilateral breast tumor recurrence, invasive contralateral breast cancer, local/regional invasive recurrence or distant recurrence) or death of any cause.
Secondary endpoints
Breast cancer-specific survival (BCSS)
Time from diagnosis/randomization to death from breast cancer. Distant recurrence-free interval (DRFI): Time from diagnosis/randomization to first documented distant recurrence or death from breast cancer. Recurrence-free interval (RFI): Time from diagnosis/randomization to first documented recurrence (invasive ipsilateral breast tumor recurrence, invasive contralateral breast cancer, local/regional invasive recurrence or distant recurrence) or death from breast cancer.
For each outcome measure, patients who did not experience any of the defined events or were lost to follow-up were censored at the date of their last contact or at 15 years after diagnosis, whichever came first. Of the 2789 AI-treated BCAC patients, 1479 (53.0%) had a delayed study entry (for 563 patients within 0.5 years after diagnosis, and for 916 patients more than 0.5 years after diagnosis). Complete DRFS data were available for 1887 patients (67.7%). Of these, 708 (37.5%) had a delayed study entry that was within 0.5 years after diagnosis for 522 patients and more than 0.5 years for 186 patients. Data for RFS were complete for 1760 of the 1887 patients (93.3%). Of these, 656 (37.3%) had a delayed study entry, that was within 0.5 years after diagnosis for 497 patients and more than 0.5 years for 159 patients.
Statistical analysis
Throughout the manuscript, statistical significance was defined as p-value < 0.05. Unless otherwise stated, all statistical tests were two-tailed.
Discovery analysis (stage 1)
For each SNP and each outcome measure, survival analysis was performed using Cox proportional hazards regression and an additive genetic model. For OS and BCSS, delayed entry Cox models were applied where time at risk was defined as starting from the date of diagnosis or date of study entry in the case of a delayed entry. For all other survival endpoints, patients entering a study more than 0.5 years after diagnosis were excluded and delayed entry was not considered. Corresponding sensitivity analyses were performed with all available patients and follow-up time, both starting from date of diagnosis and date of study entry. Analyses were conducted for all AI-treated patients as well as the subgroups of anastrozole- and letrozole-treated patients using the statistical software R (version 4.2.0; https://www.r-project.org) and additional packages rms_6.7.145 and metafor_4.4-046.
All Cox models were stratified by country and included 10 ancestry informative principal components to adjust for population structure. OncoArray and iCOGS datasets were analyzed separately and results were subsequently combined using random-effects meta-analysis. Here, standard errors of the HR estimates were recomputed based on the likelihood ratio test statistic, as previously described12. For top hits identified in the unadjusted analyses (meta-analysis p-value ≤ 1E-04), we conducted secondary analyses with adjustment for different combinations of patient characteristics (age, body mass index (BMI), menopausal status, family history of breast cancer in a first degree relative), tumor characteristics (size, nodal status, grade, stage, ER/PR status), therapy (type of AI treatment, adjuvant/neoadjuvant chemotherapy, radiation) as well as surgery and study type.
PancanQTL was examined to see whether top variants of the unadjusted analyses have been identified as cis-expression quantitative trait loci (cis-eQTL) or survival QTLs in the TCGA breast cancer cohort or other tumor entities47. In addition, the Functional Mapping and Annotation of Genome-Wide Association Studies (FUMA GWAS) tool48 was applied to check for cis-eQTLs in normal tissues including breast mammary tissue, using an eQTL p-value threshold of 1E-03. Genes identified in eQTL databases were further examined in KM Plotter for associations of their RNA-seq expression levels with overall survival in ER-positive, endocrine treated breast cancer patients with or without exclusion of patients that had received chemotherapy22.
Selection of candidate variants for validation analysis (stage 2)
The selection of candidate variants was based on the results of the discovery analysis (stage 1). For the primary endpoints (OS, DRFS, RFS), we first filtered for meta-analysis p-value ≤ 1E-04 (Supplementary Data 2, 4, 5). For the secondary endpoints (BCSS, DRFI, RFI) with less pronounced effects, a cutoff of p ≤ 5E-04 was applied (Supplementary Data 6). Selection of candidate variants based on low p-values introduces optimistic bias into the corresponding regression estimates, a phenomenon which is also known as winner’s curse bias or Beavis effect. To address this problem, we applied a bootstrap-based resampling approach for survival analysis to the discovery cohort49. We used one set of bootstrap samples (n = 500) to obtain bias-reduced HR estimates, and a second set of bootstrap samples (n = 100) to empirically estimate variance and covariance of within-sample and out-of-sample estimates for each variant. Subsequently, the power/sample size calculation for the final selection of the primary validation candidates was based on the winner’s curse-corrected estimates and a one-sided score test50. Given the number of events of the primary endpoints in the validation cohorts, the MAFs of the variants in 1000 G phase3 EUR, and a Bonferroni-corrected alpha level of 0.56% ( = 5%/9), the power was 80% to detect six selected primary candidate variants: rs3820071 (DRFS, RFS), rs6685648 (DRFS), rs10496860 (DRFS, RFS), rs3107669 (OS), rs353298 (DRFS, RFS), and rs353296 (RFS). Furthermore, as secondary candidates we selected rs11012983 and rs827423 from the anastrozole- and letrozole-only analyses, respectively, as well as rs830321, rs2276836, rs251124, rs2541311, and rs4713432 from the analyses of the secondary endpoints.
Validation analysis (stage 2)
Survival analysis in the validation cohorts was performed similar to the discovery analysis. For each validation cohort, Cox proportional hazards regression was applied to examine the associations between candidate variants and outcome measures in an additive genetic model. For OS and BCSS, delayed entry Cox models were used. For all other endpoints, patients entering a study more than 0.5 years after diagnosis were excluded from the analysis. For MA.27, Cox models were stratified by country (US, Canada) and included the previously described top seven eigenvectors of the SNP correlation matrix10. Here, survival analysis was performed separately for each of the three platforms and results were combined in a fixed-effects meta-analysis. KARMA and pKARMA patients were analyzed together, where Cox models were stratified for study. For all other validation cohorts, unstratified Cox models were applied. As described above, standard errors of the HR estimates were recomputed based on the likelihood ratio test statistic. Results of the five validation cohorts were combined in a random-effects meta-analysis. Since nearly all 643 KARMA/pKARMA patients had a delayed study entry ( > 0.5 years after diagnosis), these were only included in the OS and BCSS analysis. For all other survival endpoints, only MA.27, PreFace, MCBCS, and IKP211 were used for validation analysis.
In secondary adjusted analyses, we included nodal status and adjuvant chemotherapy as strata in the Cox models10 with and without additional adjustment for tumor size, ER/PR status, type of AI treatment (MA.27, MCBCS, and IKP211), as well as ECOG performance score and bisphosphonate use (MA.27 only). Additional stratification factors included trastuzumab as well as celecoxib and aspirin use for MA.2733. In corresponding sensitivity analyses, stratification factors were alternatively included as covariates in the Cox models. Further sensitivity analyses considered (additional) adjustment for different combinations of age or menopausal status, BMI, family history of breast cancer in a first degree relative, tumor grade, tumor stage, neoadjuvant chemotherapy, radiation, and/or surgery. All analyses were similarly performed in the subgroup of patients treated with anastrozole and letrozole, respectively.

Responses