Long-read genome sequencing resolves complex genomic rearrangements in rare genetic syndromes

Introduction
While microarrays and exome sequencing are routinely used for molecular diagnosis of genetic disorders, many laboratories are shifting to whole-genome sequencing for simultaneous detection of CNVs and small variants in both coding and non-coding regions1,2,3,4,5,6,7,8. Despite these advances in molecular testing, more than half the patients with a rare genetic condition remain undiagnosed9,10. Some of these diagnostic failures are due to the limitations of microarray and short-read sequencing in detecting structural variants and complex chromosomal rearrangements11. Single molecule long-read technologies are better equipped to address these challenges due to their ability to generate reads that are consistently larger than 10 kb12,13,14. Indeed, long-read approaches have enabled a more complete assembly of the human genome by the Telomere-to-Telomere Consortium15. Here, we resolved novel complex chromosomal rearrangements in two rare genetic syndromes using Pacific Biosciences (PacBio) long-read sequencing.
Rec8 syndrome is a rare chromosomal disorder caused by a recombination event that results in partial monosomy of terminal chr8p and partial trisomy of terminal chr8q regions16,17,18. Early reports have described typical Rec8 syndrome as a recurrent ~47.90 Mb terminal chr8q duplication recombined to the end of a ~ 11.65 Mb terminal chr8p deletion in individuals of Hispanic ancestry16,18. While most of these variants occurred de novo, they were shown to be derived from a pericentric chromosome 8 inversion in a carrier parent16,17. Characteristic features of a typical Rec8 patient include intellectual disability, congenital heart defects, seizures, and dysmorphic facial features. These previously defined spectra of rearrangements and phenotypes associated with Rec8 syndrome, however, are expanding with newer reports of atypical Rec8-like rearrangements18,19,20.
Chromosome 18q deletion syndrome is a chromosomal abnormality caused by partial deletion of the long (q) arm of chromosome 18. This syndrome is typically associated with interstitial or terminal deletions spanning several megabases21,22,23,24. The associated clinical presentations are highly variable but most often comprise developmental delay, intellectual disability, incomplete myelination, and congenital malformations23,25,26,27,28,29. Studies describing cryptic and complex chromosomal rearrangements involving chromosome 18 have identified several such rearrangements in patients with chr18q deletion. These studies, however, were unable to fully characterize the chromosomal rearrangement event that occurred28,30,31.
The two patients described herein presented with rare neurodevelopmental and craniofacial anomalies and underwent microarray testing which revealed CNVs in chromosomes 8 (patient 1) and 18 (patient 2). The genomic rearrangements associated with these CNVs could not be fully resolved by microarray analysis (or short-read genome sequencing in one patient). We therefore performed long-read genome sequencing using PacBio’s circular consensus sequencing to define two novel rearrangement events associated with these CNVs in the patients.
Results
Clinical Presentation
Patient 1 (Family 1) is a three-year-old female who is one of four children born to a non-consanguineous family of European ancestry. She was enrolled into the Congenital Craniofacial Malformations Genetics research protocol at ~1.5 years of age due to her midline facial defects, hypertelorism, nasal bridge cyst, submucosal cleft palate, agenesis of corpus callosum, subependymal gray matter heterotopia, duplicated pituitary gland, staring spells, and conductive hearing loss. After enrollment into the protocol, speech and language assessment identified delayed receptive and expressive language skills, which prompted enrollment in speech therapy. She also started to demonstrate decreased visual motor skills, fine motor precision, and hand strength, which hindered her participation in age-appropriate play activities. She exhibited impaired upper body coordination and strength and lacked spontaneous and efficient bilateral hand use. A speech pathology report at ~2.5 years of age noted incomplete laryngeal closure that led to frequent instances of laryngeal penetration progressing to silent aspiration. The family history is unremarkable with no affected parents or siblings (Fig. 1a, Left panel).

a Family pedigree for the two families in this study (arrow indicates proband), family 1 (left), family 2 (right). b Copy number change and homozygosity plots from short-read genome sequencing data show de novo chromosome 8 CNVs in family 1 (indicated by red arrows). c PacBio coverage depth plots showing de novo chromosome 18q deletions in family 2 (indicated by red arrows).
Patient 1 had a karyotype performed at birth which reported an apparent deletion involving the terminal region of the short arm of chromosome 8 (Supplementary Table 1). This finding did not rule out the possibility of a translocation of the absent chromosome 8 material and the karyotype was designated as 46,XX,?del(8)(p23.1). A Cytogenomic SNP microarray (ARUP Laboratories) was performed to further characterize the chromosome 8 abnormality seen in the karyotype. Microarray findings reported a terminal chr8p deletion of ~6.9 Mb and a terminal chr8q duplication of ~3.5 Mb (Supplementary Table 1). A terminal chr8p deletion and a terminal chr8q duplication are typically observed in Rec8 syndrome patients16,18. However, the CNVs in the patient had breakpoints that are typically not associated with a Rec8 rearrangement. The microarray report therefore suggested additional testing to clearly determine any atypical rearrangement involving these alterations. These CNVs also lacked critical genes typically noted within Rec8 CNVs, and clinical correlation was suggested for two genes (CLN8 and MCPH1) within the chr8p deletion region that are associated with autosomal recessive conditions. The family was subsequently referred to the translational research protocol to provide diagnostic clarity around the primary genetic cause of the patient’s phenotypes.
Family 1 was enrolled in the Congenital Craniofacial Malformations Genetics research study (STUDY00001876) to identify small variants within the reported chr8p deletion region that may explain the patient features. At the time of enrollment, neurodevelopmental problems that are typically associated with Rec8 syndrome patients were not appreciated in the proband, likely due to no formal assessment of developmental delays conducted at the time. The microarray report had suggested clinical correlation with two genes (CLN8 and MCPH1) within the chr8p deletion region. CLN8 is associated with autosomal recessive neuronal ceroid lipofuscinosis-8 in OMIM (MIM#600143). MCPH1 is associated with autosomal recessive primary microcephaly 1 in OMIM (MIM#3251200). We performed genome sequencing for all six individuals in family 1 using Illumina’s short-read technology to an average mean depth between 28.23x – 44.42x (Supplementary Table 2). These data were aligned to the GRCh38 reference and variants were called as described in the methods section.
Small variants were called in all individuals in the family using Churchill32. Searching for rare damaging variants led to the detection of 986 single nucleotide variants (SNVs) and indels in total. Since the proband was the only affected individual in the family, we considered variants that were consistent with autosomal recessive inheritance as well as any that occurred de novo in the proband. We found no small variants in the proband that met these criteria and thus did not identify any compelling SNVs or indels in CLN8, MCPH1 or any other gene that may explain the patient’s features.
We next performed CNV detection using VarScan2, which uses a read-depth based approach where the ratio of normalized read depth in the proband sample compared to that in normal samples are used to assign copy number states to chromosome segments33,34. We identified 186 CNVs across all 6 individuals, of which 51 were in the proband. CNVs that affect non-coding regions, were present in a healthy individual in the family, or had borderline segment ratios (absolute seg mean <0.4) were filtered out. This led to the identification of 9 de novo CNVs (2 heterozygous deletions and 7 heterozygous duplications), all which of were segments in the terminal chr8p and chr8q CNVs that were detected in the microarray analysis (Fig. 1b; Supplementary Fig. 1; Supplementary Table 3).
Since these results were indicative of a novel Rec8-like rearrangement with atypical breakpoints, we inquired about clinical features associated with Rec8 syndrome that may have been diagnosed in the proband since enrollment in the research study. Here we found formal diagnosis of developmental delays in the proband that were not noted at the time of enrollment. These additional features made a compelling case for a Rec8-like rearrangement in the proband, which led us to investigate the exact nature of any rearrangement in this patient. We therefore performed structural variant (SV) analysis to identify any such rearrangement. SVs were detected using MANTAv1.6.0 using default parameters followed by removal of predefined blacklisted SVs35. SVs that were commonly observed in the proband and a healthy individual in the family were filtered out and the resulting SV calls were analyzed using SvAnna, an SV prioritization tool that ranks SVs using a phenotype driven approach36. SvAnna is known to report any causal SV within the top 10 ranked SVs in its output. We therefore assessed the top 20 SVs that were ranked by SvAnna (Supplementary Table 4). Here we found 18 SVs that passed visual manual review but were also observed in a healthy individual despite not being called in that individual by MANTA, and two SVs that were not observed upon visual review of the aligned reads. Furthermore, any SVs linking the terminal chr8 CNVs were not detected in the MANTA output.
Investigation of the aligned short-reads on IGV, however, detected discordant reads from the chr8q duplication breakpoint mapping to low-quality mates at the chr8p23.1 deletion site (Supplementary Fig. 2a). Although this finding was indicative of a rearrangement, there were not enough breakpoint spanning reads at the chr8q duplication breakpoint (6 soft-clipped reads out of 33 total reads) in the short-read data to support recombination of a single copy of the chr8q duplicated region to the chr8p deletion site. Additionally, we could not identify any high-quality breakpoint spanning reads at the chr8p breakpoint to confirm this rearrangement in the proband. These limitations motivated us to perform long-read genome sequencing using PacBio’s HiFi circular consensus sequencing of the patient’s sample to assess any complex rearrangement in the long-read data37. HiFi mapping of the long-read data against the GRCh38 reference genome resulted in an average coverage depth of 23× for the patient sample with a mean read length of 12,618p, and a maximum read length of 41,419 bp (Supplementary Table 5, Supplementary Fig. 2b). The coverage map of the long-read sequencing data also captured the chromosome 8 copy number changes (monosomy of terminal chr8p and trisomy of terminal chr8q) previously identified in the microarray and short-read sequencing analysis (Supplementary Fig. 2c).
SVs were called from the long-read data using PacBio’s structural variant calling and analysis tool (PBSV) on SMRT Link version 13.1.0. Variants that passed the PBSV filters were compared to an internal PBSV reference dataset generated from 14 unaffected individuals. Since 14 samples were not large enough to calculate population frequencies, we filtered out any SV present in both the proband and the reference data to generate a list of unique SVs in the patient. These SVs were filtered for rarity in large population cohorts and prioritized by patient’s clinical phenotypes using SvAnna. Unilke MANTA, PBSV assigns a BND label to breakpoints representing large copy number alterations. Due to limitations of the SvAnna software in accurately detecting the underlying alterations associated with BND calls in the PBSV output, we assessed the BND calls separately. Assessment of the top 20 non-BND SVs prioritized by SvAnna identified six SVs that affected a disease-associated gene, of which only three fit a valid mode of inheritance associated with these diseases, none of which were consistent with the clinical phenotypes observed in the patient (Supplementary Table 6). We next investigated all 50 BND calls from the unique SV list and compared these against our internal reference data, which ruled out 41 BND calls that were also noted in the reference set. Of the nine unique BND calls, five were additionally found to be present in a reference sample upon IGV review that were not detected by PBSV in the reference data. The remaining four SVs (Supplementary Table 7) included (1) a translocation involving chromosomes 7 and X that did not affect any coding region and was not found to be associated with disease, (2) an ~2.75 Mb deletion on chromosome 9 with no known disease association, and larger deletions encompassing this region were reported in ClinGen and DECIPHER databases as variants of unknown significance38,39, (3) a second deletion of ~420 kb on chromosome 9 that contained only one coding gene, FAM88B, which is not known to have any disease association in the literature, and (4) a rearrangement with breakpoints at chr8:7766094 and chr8:141711312, which supported a novel Rec8-like event involving the terminal chr8 CNVs in the proband.
Review of the Rec8-like rearrangement breakpoints using IGV showed breakpoint spanning reads with supplementary alignments spanning the two chromosome 8 CNVs in the long-read data, connecting the chr8q duplication breakpoint to the end of the chr8p deletion (Fig. 2a, b). BLAT-based alignment (to GRCh38 reference genome) of 12 representative breakpoint spanning reads (out of 31 total breakpoint spanning reads) from both the chr8p deletion and the chr8q duplication breakpoints mapped these reads to a single breakpoint at the chr8q duplication breakpoint (chr8:141,711,312) and to multiple breakpoints near the chr8p deletion breakpoint with high alignment scores (>95% identity), suggesting the presence of repeat elements near the chr8p breakpoint (Supplementary Figs. 2d, e)40. This region also exhibited a relatively high read coverage in both the short-read and the long-read sequencing data compared to the rest of the genome, which was consistent across all unaffected individuals across family 1, masking the precise breakpoint location (Supplementary Fig. 3a). To better identify the repeat sequences at the chr8p deletion region, we extracted all reads around the chr8q breakpoint and mapped them to the T2T-CHM13 reference genome, which is known to better represent repetitive regions in the genome15. These reads mapped to a single breakpoint on chr8q, and to 13 breakpoints near the chr8p deletion breakpoint region within an ~200 kb window (Fig. 2c, d). Interrogation of 1500 bases up- and downstream of each of the breakpoints at the chr8p region using RepeatMasker identified a 1033 bp LTR element (LTR5A) 109 bp upstream and a 28 bp simple repeat (AAAAC)n 1452 bp downstream of each of the 13 breakpoints (Supplementary Table 8). We also detected a 77 bp LINE element (L1MB3) 1311 bp downstream of nine of these breakpoints, supporting the repetitive nature of these elements in this region. Two LINE elements (L2c and L2b) were also detected 960 bp upstream and 361 bp downstream of the chr8q breakpoint. Unearthing any possible involvement of these repeat elements in the rearrangement will require additional work beyond the scope of this manuscript. The chr8p deletion was supported by the apparent change in sequencing coverage in the proband sample before and after this repetitive region (Fig. 2f). Coverage tracks of the long-read alignments exhibit a drop in coverage to the left of the repeat region in the proband in both the short and the long-read coverage data, representative of a heterozygous deletion, which reverts to diploid coverage as we move past this region. This change in coverage is absent from the unaffected individuals in the family (Supplementary Fig. 3a). The duplication breakpoint is also supported by a change in coverage, where the coverage increases at the duplication breakpoint, clearly defining the duplication breakpoint to a base pair resolution at the genomic location chr8:141,711,312 (GRCh38) in the proband alone (Fig. 2e; Supplementary Fig. 3b).
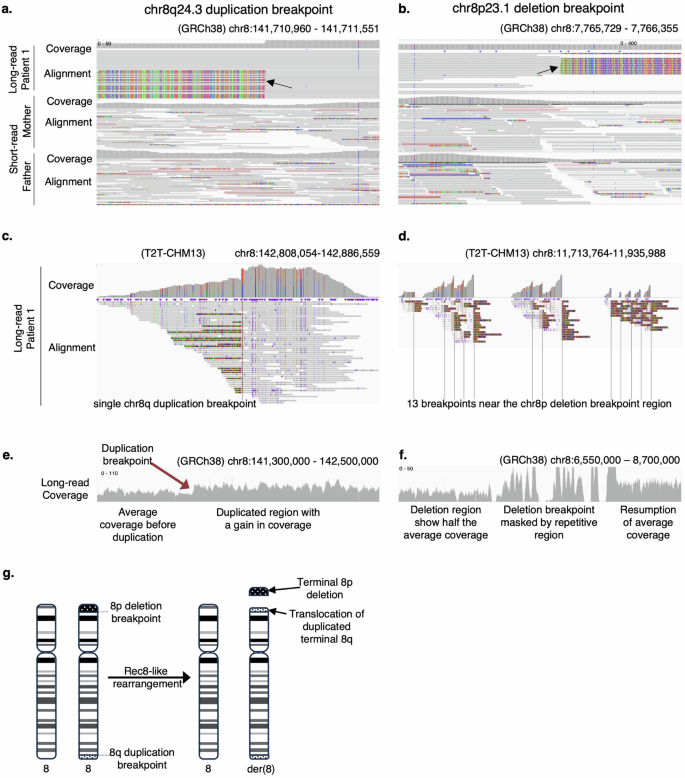
IGV view of total coverage and read alignment of PacBio circular consensus sequencing reads at chr8q duplication breakpoint, sorted by parental haplotypes (a) and the chr8p deletion breakpoint, sorted by breakpoint spanning reads (b). Colored portion of the soft-clipped (breakpoint spanning) reads shows base mismatches that spans the rearrangement. Only reads with mapping quality >30 were considered for analysis. IGV view of minimap2 alignment of chr8q breakpoint spanning reads to the T2T reference genome maps to chr8q duplication breakpoint (c) and the chr8p deletion breakpoint region (d). IGV view of long-read coverage at the chr8q duplication region (e) and the chr8p deletion region (f). g Schematic of the rec-8-like rearrangement in the proband in family 1.
The chr8p deletion region and the chr8q duplication region together encompass 206 genes in total. Twenty-two of these are associated with a disease in OMIM, none of which independently explained the proband’s condition (Supplementary Table 9). Literature reports of Rec8 syndrome patients showed that the typical Rec8 rearrangement has a history of occurring in individuals of Hispanic ancestry and are associated with a balanced inversion event within chromosome 8, inherited from an unaffected parent16,17. Analysis of the long-read data at the chr8q duplication breakpoint showed 17 out of 44 total reads as breakpoint spanning reads that map the chr8q duplication to the chr8p deletion region, showing about one-third of the reads connecting the two CNVs. In the case of an inversion associated with the terminal CNVs observed in this patient, we would expect about one-third of the reads to show an inversion event at this position as well, which was not present in our long-read sequencing data. We next sorted the reads near the chr8q breakpoint into parental haplotypes based on a heterozygous SNP at position (GRCh38) chr8:141711508 in the patient that was homozygous in the father (Fig. 2a). This approach traced the affected allele (with the soft-clipped reads) in the proband to the unaffected mother. Investigation of this breakpoint in the proband’s long read and the mother’s short read data did not provide any support for an inversion event. In fact, no breakpoint spanning reads supporting an inversion were observed in either of the parents or any of the sisters (Supplementary Fig. 4). Given the Hispanic ancestry associated with an inversion around the typical Rec8 breakpoints, it is likely that the atypical rearrangement in the patient occurred de novo without any prior inversion event in an unaffected parent, in part, due to her non-Hispanic (European) ancestry. While attempts at PCR amplification and Sanger confirmation of this rearrangement failed (Supplementary Fig. 5; Supplementary Table 10), likely due to the repetitive nature of the chr8p breakpoint, long-read data was successful in validating the terminal chromosome 8 CNVs and identifying this novel Rec8-like rearrangement in the patient. Our findings therefore provided diagnostic clarity for the clinicians and the patient’s family and identified a novel Rec8-like rearrangement in a patient of non-Hispanic ancestry (Fig. 2g).
Patient 2 (Family 2) is a two-year-old female who presented with global delays, hypotonia, and dysmorphic features including brachycephalic head shape, slightly low set ears, thickened helices, bilateral ear lobe pits, flat nasal bridge, broad and bulbous nose, deep-set eyes, and mild hypotelorism. She had a history of bradycardia and respiratory distress after delivery, and poor weight gain at six months of age. Her global delay included delayed motor and speech development. Her receptive language was more intact than her expressive language. At two years and six months of age, she spoke about 10 words but did not use them together. Her hearing and vision were normal. She started rolling at five months of age, sat at 11 months, crawled at 14 months, and started walking at two years and four months. She has been in physical therapy since she was nine months old, and occupational and speech therapy since she was two years old. Linear growth had been on the slower end of normal, tracking at the 8th percentile for height. Weight and head circumference were normal. Head MRI findings at one year and ten months of age showed patchy T2 hyperintensities in the subcortical white matter, which were nonspecific and could represent gliotic changes from old insult. There were no concerns about seizures. Chest X-rays performed in the setting of acute respiratory symptoms were normal with no congenital malformations or cardiopulmonary findings. With normal growth and no murmur on examination, cardiac malformation was unlikely, and echocardiogram was not performed. She is the only child of a non-consanguineous family of European ancestry with no similar features reported in her parents (Fig. 1a, Right panel).
SNP microarray was performed for patient 2 at 14 months of age to detect large chromosomal abnormalities. Microarray analysis reported three deletions at chr18q11.2-q12.2 (deletion 1), chr18q12.3 (deletion 2), and chr18q21.1 (deletion 3), respectively (Supplementary Table 1). Interstitial and terminal deletions of variable sizes and breakpoints on the long arm of chromosome 18 are associated with autosomal dominant chromosome 18q deletion syndrome21,22,23,24. This syndrome is characterized by highly variable clinical features, the most common of which are short stature, intellectual disability, hypotonia, hearing impairment, and foot deformities. The three interstitial deletions in the patient that are indicative of a complex rearrangement have not been previously reported in a patient with chromosome 18q deletion syndrome. The patient’s parents wanted to have another child and were interested in finding out about any risk of recurrence during future pregnancies. Screening the parents was therefore important to identify any structural variants that could explain the deletions observed in the proband. Extended peripheral blood chromosome analysis and fluorescent in situ hybridization analysis were inadequate in identifying any chromosomal rearrangement in the proband. These assays also did not identify any abnormalities in the parents, suggesting a de novo occurrence of these chr18q deletions. The family was enrolled in research to resolve any chromosomal rearrangement in the patient and to identify any alterations in the unaffected parents, which would provide diagnostic clarity and identify any possible carrier alterations in the unaffected parents that may speak toward a risk of recurrence in future pregnancies.
We consented the proband and her parents under the Rare Diseases/Genome Sequencing research protocol (IRB:11-00215) for genomic testing. We performed PacBio circular consensus sequencing on genomic DNA from all three individuals to resolve the potential rearrangement indicated by the three proximal deletions on chromosome 18. An average coverage depth of 20–27× was achieved in the long-read experiment with mean read lengths between 12,335 bp and 13,613 bp, and maximum read lengths ranging from 32,282 bp to 50,563 bp (Supplementary Table 5, Supplementary Fig. 6). HiFi mapping and PBSV analysis were run on SMRT Link using default parameters against the GRCh38 reference genome. Coverage maps generated from HiFi mapping of the long-read data successfully identified the three chr18q deletions in the patient but not in her parents (Fig. 1c).
A systematic approach at filtering and prioritization of non-BND SV calls from the PBSV output (as described for patient 1) identified three non-BND SVs within the top 20 SvAnna prioritized SVs that affected a coding transcript, of which only two were found to be associated with disease, neither of which explained the proband’s clinical phenotypes and were ruled out from further assessment (Supplementary Table 11). The BND calls from the patient sample were separated from the PBSV output and compared against the BND calls in the internal reference dataset. Out of 49 BND calls, 41 were commonly found in the reference dataset, another 4 were identified in the reference data upon IGV review that were not called by the PBSV algorithm in the reference samples. The remaining 4 BND calls represented a single structural variant that involved the three interstitial deletions identified in the microarray as well as two additional breakpoints at positions chr18:49306872 and chr18: 49306881 (Supplementary Table 12). Assessment of these breakpoints on IGV identified breakpoint spanning reads at all six breakpoints of the three reported deletions, which were supported by an apparent drop in coverage at the deletion sites and allowed precise mapping of the deletion breakpoints to a single base pair resolution (Fig. 3a, Supplementary Fig. 7). Investigation of the two additional breakpoints revealed a fourth deletion of 9 bp around an inversion site located 1.17 Mb downstream of deletion 3 (Fig. 3b).

a IGV view of the left breakpoint (left panel) and the right breakpoint (right panel) of deletion 1 at chr18q11.2-q12.2 in family 2. b IGV view of aligned PacBio circular consensus reads at the inversion breakpoint 1.174 Mb downstream of deletion 3 (left panel) and the zoomed-in view of the 9 bp deletion at the inversion breakpoint (right panel). Breakpoints from deletions 2 and 3 are shown in Supplementary Fig. 7. Long reads in the patient were grouped by SNPs inherited from a parent (indicated by arrows) in order to differentiate the parental haplotypes in the proband.
The BND calls from the PBSV output, and BLAT based alignment of a representative breakpoint spanning read from each breakpoint was in concordance about the nature of the rearrangement that was identified as shown in Fig. 4a. The inversion breakpoint is precisely mapped between chr18:49,306,873 −49,306,881 with the 9 bp deletion spanned by two types of breakpoint-spanning reads. The breakpoint spanning read on the left side of the inversion mapped to the reverse strand at the right breakpoint of deletion 2. The breakpoint spanning read on the right side of the inversion mapped to the forward strand at the right breakpoint of deletion 1. Breakpoint spanning read from deletion 1 mapped the left side of deletion 1 to the right side of deletion 3, and the right side of deletion 1 to the inversion breakpoint. The left side of deletion 2 mapped to the reverse strand on the left side of deletion 3, and the right side of deletion 2 mapped to the inversion breakpoint. The left side of deletion 3 mapped to the reverse strand on the left side of deletion 2, and the right side of deletion 3 mapped to the left side of deletion 1. A schematic of the resultant derivative chromosome 18 is depicted in Fig. 4b. Deletion 1 was precisely mapped to chr18:23,614,482–36,003,481 resulting in a 12,388,999 bp heterozygous deletion. Deletion 2 was precisely mapped to chr18:39,652,549–41,450,560, resulting in a 1,798,011 bp heterozygous deletion. Deletion 3 was precisely mapped to chr18:46,795,9421–48,132,510, resulting in a 1,336,569 bp deletion (Supplementary Table 13). RepeatMasker analysis of 1500 bases up- and downstream of these breakpoints identified several LINE, LTR, and Alu elements that may play a role in the driving this rearrangement (Supplementary Table 14). Further investigation is therefore necessary to clearly define any role of these repeat elements in the chromosomal rearrangement in the patient, which is beyond the scope of this study. Grouping the long-reads in the patient by SNPs inherited from one of the parents allowed sorting of these reads into parental haplotypes. These data demonstrated that the affected allele in the proband was inherited from the unaffected father (Fig. 3a, b; Supplementary Fig. 7). Investigation of the rearrangement breakpoints in the unaffected father (and the mother) did not show any breakpoint spanning reads indicative of any carrier alterations (Fig. 3 and Supplementary Fig. 7). Moreover, PCR amplification and Sanger sequencing across these breakpoints confirmed the rearrangement in the patient, but not in her parents (Supplementary Figs. 8, 9; Supplementary Table 10). Together, these data support a de novo occurrence of this previously unreported rearrangement in the patient that provided diagnostic clarity and suggested a low risk of recurrence in future pregnancies, which was a significant concern for this family.

a USCS Genome Browser view of BLAT alignments of the representative breakpoint spanning reads. Track 1 shows the breakpoints of three deletions and one inversion with a 9 bp loss on chromosome 18q. Track 2 shows the chr18 segments retained, with colored arrows (in color spectrum order) to indicate how they were assembled into a derivative chromosome. Solid lines indicate preserved strands; dashed lines indicate strand inversion. Tracks 3 through 10 show the alignments of representative breakpoint-spanning reads. Each read has two high-scoring alignments: one to the region it came from, and one to a different breakpoint. Deletion 1 breakpoints (left = BP1, right = BP2); Deletion 2 breakpoints (left = BP3, right = BP4); deletion 3 breakpoints (left = BP5, right = BP6); Inversion breakpoint = BP7. BP1 read maps to BP6, BP2 read maps to BP7, BP3 read maps to BP5 on the reverse strand, BP4 read maps to BP7, BP5 read maps to BP3 on the reverse strand, and BP6 read maps to BP1. b A schematic of the genomic algebra behind the chr18q rearrangement in derivative chromosome 18 [der(18)]. Retained segments are labeled A through E.
Discussion
In this study, we employed PacBio’s HiFi circular consensus sequencing to resolve previously unreported complex rearrangements underlying rare genetic conditions in two unrelated individuals. In patient 1, we identified two copy number changes at the p- and q- termini of chromosome 8 in the proband who harbored a novel Rec8-like chromosomal rearrangement. Analysis of publicly available datasets for chr8p deletions and chr8q duplications identified several similarly sized or overlapping deletions and duplications in patients with variable phenotypes that were deemed pathogenic or likely pathogenic in DECIPHER (Supplementary Table 15)38. While some of these reported features in DECIPHER patients were present in patient 1, they did not fully explain the patient’s condition. Long-read genome sequencing resolved these CNVs in the proband as a novel Rec8-like rearrangement that was not reported previously in the literature. Several publications have described patients with partial monosomy chr8p/trisomy chr8q of varying sizes that were associated with Rec8 syndrome16,17,18,19,20,41. Rec8 syndrome is described in OMIM as a chromosomal disorder with affected individuals typically presenting with intellectual disability, congenital heart defects, seizures, and craniofacial features (MIM#179613). Cohort-level interrogation of 65 Rec8 syndrome patients in Sujansky et al. described common abnormalities associated with Rec8 syndrome16; many of these features overlapped with features in patient 1 in this study: developmental delays (present in 100% of Rec(8) patients), hypertelorism (100%), hearing loss (73%), seizures (50%), cleft lip and/or palate (29%), agenesis of corpus callosum (7%). Although the authors of the study identified congenital heart disease as a major malformation in Rec8 syndrome patients (present in 93% of Rec8 patients), patient 1’s echocardiogram did not reveal any cardiac abnormalities. Stevens et al. had associated cardiac abnormalities in their Rec8 syndrome patient with haploinsufficiency of the GATA4 transcription factor that resided within the chr8p monosomy region in their study19. Additionally, Habhab et al. mapped cardiac anomalies to the duplication segment between chr8q22.2 and chr8q23.1. They noted that the cardiogenic genes located within this chr8q duplicated locus may be responsible for cardiac defects in their patient with Rec8 syndrome who lacks loss of the chr8p segment that contains GATA418. It is, therefore, likely that the lack of copy number alterations in patient 1 affecting these cardiogenic genes explain the lack of cardiac abnormalities in the patient.
The typical rearrangement reported in Rec8 syndrome have a duplication of chr8q (q22 → qter) and deletion of chr8p (pter → p23), which are larger than the CNVs observed in our case. “Classic” Rec8 syndrome is associated with a ~ 11.65 Mb chr8p deletion (chr8pter → chr8p23.1) and a ~ 47.9 Mb chr8q duplication (chr8q21.11 → chr8qter). However, several reports since then have identified atypical rearrangements of varying sizes in patients with Rec8 syndrome. Vera-Carbonell et al. described a female patient with a ~ 2.27 Mb deletion of chr8p (chr8pter → chr8p23.2) and a ~ 41.93 Mb duplication of chr8q (chr8q22.3 → chr8qter) who presented with a spectrum of dysmorphic features, generalized hypertonia, and delayed psychomotor development20. Stevens et al. described a male patient with a 16.9 Mb chr8p deletion (chr8pter → chr8p22) and a 21.7 Mb chr8q duplication (chr8q24.13 → chr8qter) who presented with cardiac malformations, dysmorphic facial features, hypospadias, cryptorchidism, intellectual disability, speech delay, motor delay, and feeding problems19. Habhab et al. described a fetus with Rec8 syndrome that presented with intrauterine growth restriction, hypogenesis of corpus callosum, bilateral cleft lip and palate, and congenital heart defect with a 1.4 Mb terminal copy number loss of chr8p (8pter → 8p23.2) and a 69.8 Mb terminal copy number gain of chr8q (chr8q21.11 → chr8qter). Our study reports a novel atypical rearrangement in patient 1 that have marked overlap of clinical features with those described for Rec8 syndrome but has a much smaller chromosome 8q duplication compared to the typical as well as the atypical Rec8 rearrangements that have been reported in the literature thus far (3.43 Mb compared to >40 Mb consistently reported in Rec8 patients). Although there is evidence of a chromosome 8 inversion in a carrier parent in classic Rec8 syndrome cases16,17, we do not observe such an inversion event in our long-read sequencing data of the proband sample; neither is such an inversion event apparent in any of the healthy individuals in the family in the short-read sequencing data. Given the atypical breakpoints for the Rec8-like rearrangement and the non-Hispanic ancestry of patient 1, it is likely that this rearrangement occurs without inheritance of a prior pericentric inversion of chromosome 8 from an unaffected parent. Formal classification of this rearrangement as a Rec8 rearrangement will therefore expand the phenotypic spectrum as well as the range of chromosome 8 rearrangements observed in Rec8 syndrome patients.
In family 2, we described a de novo chromosomal rearrangement connecting four deletions in the proband, one of which was a 9 bp deletion around an inversion site. Although this exact rearrangement is unreported in the literature, it partially overlaps with deletions commonly observed in chromosome 18q deletion syndrome patients. Independent evaluation of the four deletions in patient 2 identified several pathogenic/likely pathogenic deletions of different sizes and locations in DECIPHER that were associated with varying clinical features (Supplementary Table 16)38. Common clinical presentations include short stature, developmental delays, and congenital malformations. Genes associated with diseases in these regions are summarized in Supplementary Table 13 and Supplementary Table 17. Of note, the ASXL3 gene was associated with autosomal dominant Bainbridge-Ropers syndrome, which is characterized by developmental delays, intellectual disability, hypotonia, feeding difficulties, and dysmorphic facial features42.
Several studies have mapped specific segments on the long arm of chromosome 18 to specific phenotypes. Soileau et al. associated short stature to three different regions on chr18q: 18q12.1-12.3, 18q21.1-q21.33, and 18q22.3-q23 and noted GALR1 as a likely gene involved in growth delays29. While patient 2 was reported to have poor weight gain at six months of age, GARL1 did not overlap the chromosome 18 rearrangement in patient 2. However, a partial overlap of the patient 2 rearrangement with the chr18q12.1 and the chr18q21.1 regions with known growth defect association may provide some clues about genes other than GALR1 that maybe associated with growth delays in patients with chromosome 18q rearrangements.
Clinical features like microcephaly, cleft lip and cleft palate, IgA deficiency, renal dysplasia, congenital aural atresia, and hearing impairment that are attributed to regions outside of the chr18q rearrangement described in this study were not reported in patient 229,43. We also identified several genes within the chr18q rearrangement that fit mode of disease inheritance but appear to be incompletely penetrant in patient 2 (Supplementary Table 8). SMAD2 and DSC2 located in deletion 1 are associated with autosomal dominant conditions with cardiac phenotypes in OMIM: (i) Congenital heart defects with or without heterotaxy (MIM#619656), (ii) Loeys-Dietz syndrome 6 (MIM#619656), and (iii) Arrhythmogenic right ventricular dysplasia 11 (MIM#610476). Although cardiac defects are not apparent in patient 2, evaluation of cardiac defects in patients with similar deletions is recommended to rule out cardiac defects associated with variants in these genes. The inversion site in the chromosome 18q rearrangement in patient 2 overlaps with DYM, a gene that is associated with an autosomal recessive condition characterized by skeletal anomalies. These features were also not appreciated in the proband, likely due to a heterozygous variant in this gene where disease-causing variants are known to be homozygous.
Deletions of chr18q have been previously associated with other complex rearrangements28,30,44,45. Gruchy et al. described a patient with a t(5;18) translocation, while Rizzolio et al. reported a balanced translocation of t(X;18), proposing an association of the translocation with growth delays through distal regulation of a short stature critical region30,45. Brkanac et al. identified five patients with complex cryptic rearrangements that were not detectable by common methodologies28. These alterations can impact the phenotypes in patients, given the location and nature of the variant. Since clinical microarrays are unable to detect the extent of complex rearrangements that maybe associated with chr18q deletions, many such rearrangements likely go unnoticed in patients formally diagnosed with chromosome 18q deletion syndrome. Additional testing, including long-read genome sequencing, could therefore be considered for the identification of any cryptic rearrangement associated with a chr18q deletion finding, especially in cases where the deletion by itself may not fully explain the patient features.
Considering the limitations of microarray and short-read sequencing in resolving complex chromosomal rearrangements along with the decreasing cost of long-read genome sequencing, a compelling case can be made for more mainstream use of long-read technologies to improve the diagnostic yield in patients with rare genetic disorders. This approach may also serve to test for carrier alterations in parents and thereby better define the risk of recurrence of these complex rearrangements in future pregnancies.
Methods
Study participants and enrollment
Our study included patients who were referred by their clinicians after presenting a phenotype consistent with a rare genetic disorder. Two unrelated families exhibiting craniofacial and neurodevelopmental phenotypes with prior microarray findings were enrolled as part of Institutional Review Board (IRB) approved studies within the Steve and Cindy Rasmussen Institute for Genomic Medicine at Nationwide Children’s Hospital, Columbus, Ohio. Genomic analysis was performed on DNA isolated from the probands’ and their family members’ peripheral blood or saliva samples.
Ethics statement
This study was performed in accordance with the ethical standards of the Declaration of Helsinki and was approved by the Institutional Review Board (IRB) within the Steve and Cindy Rasmussen Institute for Genomic Medicine at Nationwide Children’s Hospital, Columbus, Ohio. Family 1 was enrolled in STUDY00001876: Congenital Craniofacial Malformations Genetics. Family 2 was enrolled in IRB:11-00215: Rare Diseases/Genome Sequencing.
Consent to publish
Informed consent was obtained from all study participants for enrollment into the IRB-approved studies, which includes consent for publication of results without revealing the participant’s identity. For pediatric patients, written informed consent was obtained from a parent/guardian to participate and publish the study.
Microarray analysis
For the proband in family 1, Cytogenomic SNP microarray was performed on cord blood using the CytoScan™ HD Suite (Thermo Fisher Scientific) according to an undisclosed, validated protocols at ARUP Laboratories. The CytoScan HD array contains ~2.67 million markers across the genome with one probe every 1150 bp. Generally, copy number changes reported by ARUP laboratories include losses greater than 50,000 bp and gains greater than 400,000 bp. Genomic coordinates correspond to the Genome Reference Consortium human reference genome build 37 (GRCh37).
For the proband in family 2 comparative genomic hybridization (CGH) microarray analysis was performed at The Institute for Genomic Medicine at Nationwide Children’s Hospital, Columbus, Ohio, using a custom-designed Agilent 180k CGH + SNP array targeted to specific regions. Array comparative genomic hybridization was used for copy number analysis. This array consists of ~120,000 probes with an average spacing of one probe per 25,000 bp throughout the genome and one probe per 10,000 bp in regions of known clinical significance. Copy number calls were based on ~5 contiguous probes reaching significance levels (log ratio < −0.5 OR > 0.5) and is not a reliable tool to detect low-level mosaicism or genomic imbalances less than ~100,000 bp in size. Genomic coordinates correspond to the Genome Reference Consortium human reference genome build 37 (GRCh37). Data interpretation was performed using GENA analysis tool by Sivotec Bioinformatics (https://www.sivotecbioinformatics.com/)
Illumina short-read genome sequencing and analysis
Genome sequencing was initially performed for six members of family 1, including the proband (patient 1), unaffected mother, unaffected father, and three unaffected sisters. Library preparation was done using NEBNext Ultra II (New England Biolabs) and paired-end sequencing (2 × 150 bp) was performed using an Illumina NovaSeq 6000 instrument according to manufacturer protocols (Illumina Inc.). Reads were aligned to the GRCh38 reference sequence, and secondary data analysis was performed using Churchill32, which implements the Genome Analysis Toolkit (GATK) “best practices workflow” for alignment, variant discovery, and genotyping. We generated ∼105 Gbp of uniquely mapped reads per individual, achieving ∼34× haploid coverage on average (Supplementary Table 2).
Our general approach to variant annotation and prioritization has been described previously46. Variants were called in all family members using GATK v.4.0.5.1, and the resulting VCF file was annotated with genes, transcripts, function classes, damaging scores, and population allele frequencies using an in-house pipeline built around the SNPeff annotation tool47. After removing common variants with minor allele frequency (MAF) > 0.01 in gnomAD (v4.1.0), we selected all splice site, frameshift, and nonsense variants for further analysis. Missense variants predicted to be damaging by 75% of in silico pathogenicity prediction tools (including REVEL, MetaRNN, CADD, SIFT, Polyphen2) were also included. A minimum quality filter was applied to filter out low-quality variant calls (QUAL > 120). The resulting variants were assessed for mode of inheritance, disease association and correlation with the patient’s clinical phenotypes.
Copy number variants (CNVs) were identified using VarScan 2 with default parameters33,34. VarScan 2 calculates and normalizes read depths to account for variations in overall sequencing depth and genomic coverage between samples. It then segments the genome into regions that are likely to have consistent copy number. Once segments are defined, VarScan2 uses a comparison of normalized read depths between the proband and normal samples to infer copy number changes. Each segment is assigned a copy number state based on the ratio of proband sample read depth to normal read depth. We applied default thresholds to these ratios to classify the segments into different copy number states—gain, loss, or normal copy number. The final output was assessed for CNVs that affected coding transcripts and/or were associated with a disease phenotype in the literature.
Structural variants (SVs) were identified using MANTA v1.6.0 with default parameters35. Here we used the primary chromosomes of GRCh38, excluding ALT assemblies, HLA regions, and other minor contigs. Notable problematic regions of the genome were excluded via a blacklist regions bedfile that was created using the ENCODE genomic regions blacklist (Amemiya et al., 2019), the GRIDSS’s (Cameron et al., 2017) SV detection blacklist, and telomeric and centromeric regions that had not otherwise been excluded. SVs that appeared in a healthy individual in the family were filtered out from the patient samples using BCFtools48. The resulting SVs unique to the proband were prioritized using SvAnna36, an SV filtering and prioritization tool that ranks SVs based on the patient’s phenotypic presentation. SvAnna was used to filter out all SVs supported by less than 4 reads and those that overlapped at least 75% of a known SV present in large population databases at >0.01% frequency (Database of Genomic Variants, GnomAD SV, Human Genome Structural Variation Consortium SVs freeze 3, and dbSNP v151). SvAnna ranks each SV based on predicted effect of the variant on gene function and matching a list of patients HPO terms to disease models on HPO. This phenotype driven approach assigns a pathogenicity score to each SV and has been shown to rank any disease-causing SV at the top of the list. We therefore assessed the breakpoints for the top 20 SVs from this ranked list using IGV and determined pathogenic variants based on mode of inheritance, disease association in the literature, and correlation with the patient’s clinical phenotypes.
Pacific Biosciences (PacBio) long-read genomic DNA and SMRTbell library preparation
Blood samples from the enrolled individuals were processed to peripheral blood mononuclear cells using a SepMate/Lymphoprep density gradient separation. Genomic DNA (gDNA) was then extracted using QIAGEN’s AllPrep Mini Kit. The quantity and quality of gDNA were evaluated using the Thermo Fisher Scientific Qubit 1x dsDNA HS Assay Kit and the Agilent TapeStation 4200 Genomic ScreenTape. The gDNA extracted from the proband in family 1 (3000 ng input) was sheared using the Megaruptor 3 (Diagenode, Liege, Belgium) according to the manufacturer’s protocol, aiming for a target mode size of 20,000 bp. A PacBio gDNA – high fidelity (HiFi) – SMRTbell library was then generated using the PacBio HiFi Express Template Prep Kit 3.0 protocol (PN 102-166-600, April 2022), utilizing 2800 ng of the sheared gDNA. In the family 2 trio, the proband’s genomic DNA (4000 ng input) underwent three passes through the Megaruptor 3 to achieve a 25,000 bp mode size. The mode size of parental gDNAs from family 2 was already suitable for library construction (size of ~24,000 bp) and did not require mechanical shearing. Subsequently, 4000 ng of each parental gDNA and 3300 ng of sheared proband gDNA were used as input for the PacBio HiFi Express Template Prep Kit 3.0 to prepare PacBio gDNA HiFi SMRTbell libraries. Each final SMRTbell library was subjected to size-selection using PacBio AMPure PB Beads (PN 100-265-900) to remove molecules smaller than 5000 bp. The size-selected libraries were assessed for DNA concentration using High Sensitivity Qubit (Thermo Fisher Scientific, Waltham, MA) and size determination using Genomic ScreenTape (Agilent, Santa Clara, CA). The resulting sample data was then imported into the PacBio SMRT Link Sample Setup (version 11.1.0.166339) to determine a per-sample complexing protocol using the Sequel II Binding Kit 3.2.
PacBio circular consensus sequencing and data analysis
Each complex library was sequenced on two SQIIe 8M SMRT Cells using adaptive loading with an on-plate loading concentration of 55 pM. The sequencing process included a 2-h pre-extension and a 30-h movie collection time. Primary analysis of the sequencing data utilized the PacBio HiFi Mapping application within SMRT Analysis using default parameters for alignment to reference build GRCh38. This aims to determine the mean coverage of sequencing depth and to generate aligned BAM files for subsequent variant interpretation. A mean coverage of 23× for the proband in family 1, 20× coverage for the proband in family 2, and 27× coverage for each of the parental samples in family 2 were obtained. Structural variants were identified using PacBio’s PBSV pipeline on SMRT Link v13.1.0 using default analysis parameters. We also generated an internal PBSV reference dataset from PacBio HiFi circular consensus sequencing of DNA samples from 14 unaffected individuals—7 from our Rare Diseases/Genome Sequencing translational research protocol, and 7 from the NIGMS Human Genetic Cell Repository at the Coriell Institute for Medical Research: NA24149, NA24143, HG03736, HG00268, HG00514, HG00733, and NA19240.
SV calls from PBSV that appeared in the internal reference data were filtered out from the patient samples using BCFtools and the resulting SVs unique to the proband were prioritized using SvAnna36 as described above. Unlike MANTA, PBSV assigns BND calls to large chromosomal events, and the underlying alteration(s) associated with these BNDs are not detectable by SvAnna when performing prioritization. We therefore separated all BND calls from the patient samples and compared them against the BND calls in our internal reference dataset to filter out any BND calls in the patient sample that are also observed in the reference data. Any unique BND call present in the patient was individually assessed using IGV. Pathogenicity of the remaining BND calls was assessed based on their predicted impact on gene function, valid modes of inheritance, and phenotypic associations with known pathogenic SVs in the literature or OMIM. The top 20 non-BND SVs from the SvAnna output were assessed as described above.
Extraction of GRCh38 mapped long reads from the chr8 duplication breakpoint in patient 1 was performed using SAMtools v1.1948. Realignment of these long breakpoint reads to the T2T-CHM13 (v2.0) reference genome was performed using minimap249,50. Repeat analysis of each rearrangement breakpoint region was performed using RepeatMaskerv4.0.9 with RMBLASTv2.14.1+. Targeted analysis of long-read sequencing was performed in Integrative genomic viewer (IGV) version 2.16.0. Breakpoint spanning reads in the proband samples were separated into parental haplotypes in IGV by sorting the reads by a nearby base position harboring a single nucleotide variant inherited from one of the parents.
PCR amplification and Sanger sequencing
PCR amplification of the region of interest was performed using the Platinum™ SuperFi II PCR Master Mix (Invitrogen, #12368010). Primer sequences used for the reactions are listed in Supplementary Table 10. Supplementary Fig. 5 and Supplementary Table 7 are schematics showing the primer design strategy across each rearrangement breakpoint for Patient 1. Supplementary Figs. 8 and 9 show the schematic of the rearrangement, primer designs, and Sanger results for Patient 2. The PCR conditions were as follows: 98 °C 30 s, 35 × (98°C 5 s, 60°C 30 s, 72°C 45 s), 72°C 5 min, 4 °C hold. PCR amplification was followed by the purification of the PCR products using the QIAquick Purification Kit (QIAGEN), which was evaluated on Agilent 4200 TapeStation per manufacturer’s protocol. Forward and reverse sequencing reactions were performed with the Big Dye v3.1 terminator mix (Thermo Fisher Scientific) and M13-tagged primers. Sequencing was performed on an Applied Biosystems 3730 instrument (Thermo Fisher Scientific). We analyzed the electropherogram results using Sequencher v5.4.6.


Responses