Metabolic reprogramming in hepatocellular carcinoma: mechanisms and therapeutic implications
Introduction
Metabolic rewiring in cancer entails adaptive modifications to meet specific demands imposed by enhanced proliferation. This rewiring encompasses both anabolic and catabolic pathways, enabling cells to maintain uncontrolled replication and, ultimately, survival1.
The liver, as the primary metabolic organ, plays an important role in regulating metabolic processes essential for overall homeostasis. These include glucose, protein and lipid metabolism, which impact energy balance regulation, nutrient storage, detoxification and the synthesis of vital biomolecules2,3,4. Hepatocellular carcinoma (HCC), often detected late, presents diagnostic challenges due to it frequently occurring alongside pre-existing liver conditions such as cirrhosis, chronic hepatitis B or C infection, or metabolic dysfunction-associated fatty liver disease. This complicates treatment and limits therapeutic options. Moreover, HCC exhibits resistance to certain chemotherapeutic agents and is prone to high recurrence rates post-treatment. The liver’s unique metabolic and detoxifying capabilities also influence the pharmacokinetics of drugs, further complicating treatment. Despite the FDA approval of the first tyrosine kinase inhibitor (TKI) for HCC treatment, resistance develops rapidly in many patients5,6. Similarly, other TKIs have shown limited survival benefits7,8. Nivolumab, an FDA-approved immune checkpoint inhibitor for second-line treatment, has shown limited efficacy in patients with HCC, further underscoring the urgent need for better therapeutic strategies9. Additionally, the liver’s susceptibility to metastases from primary cancers in the gastrointestinal tract poses further challenges10,11. This review provides current insights into cancer metabolism, specifically within the liver, and discusses potential therapeutic approaches that exploit metabolic pathways altered in HCC to inhibit tumor growth.
Main
Enhancement of glycolysis
The liver plays a central role in the regulation of blood glucose levels. The body stores glucose in the form of glycogen during periods of surplus and subsequently releases glucose into the circulatory system when blood glucose levels decrease, thereby mediating glucose homeostasis. During periods of fasting or heightened energy requirements, the liver releases glucose via glycogen catabolism4. In HCC, there is a marked reliance on glycolysis for energy production, even in the presence of oxygen, a phenomenon known as the Warburg effect12. This shift from oxidative phosphorylation to glycolysis allows HCC cells to efficiently produce energy in oxygen-rich environments, typical of anaerobic metabolism. Key glycolytic genes, including those encoding glucose transporters (GLUTs), hexokinase (HK) and pyruvate kinase (PK), are differentially expressed in HCC, leading to increased glucose uptake and metabolism (Fig. 1).
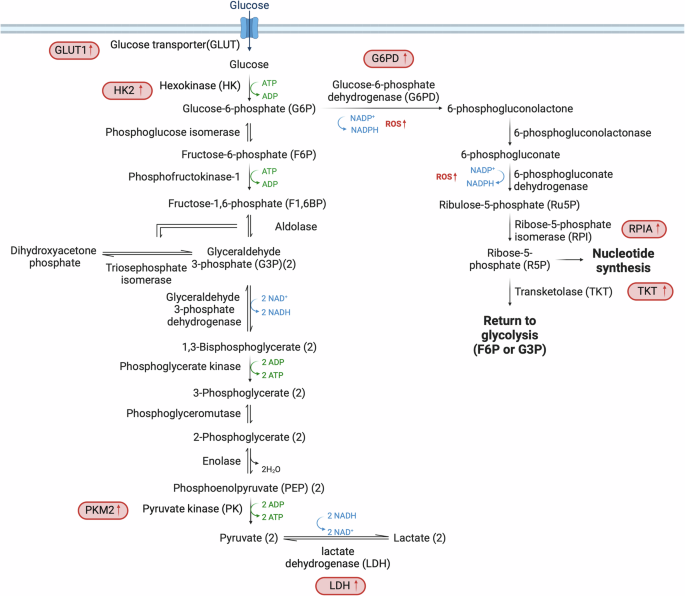
The significant shift in glucose metabolism within HCC cells, characterized by upregulated expression of key enzymes that enhance glucose uptake and utilization. These changes include increased levels of GLUT1 and HK2, which elevate glucose uptake; PKM2, which increases augments pyruvate formation; and LDH, which facilitates the conversion of pyruvate to lactate, supporting anaerobic glycolysis. The diagram also highlights the activation of the PPP, a crucial metabolic pathway that provides R5P for nucleotide synthesis and NADPH for lipid synthesis and antioxidant defense. In HCC, the PPP is often enhanced, as indicated by increased activity of enzymes such as G6PD and TKT. Enzymes with increased expression in HCC are marked in red, denoting their upregulation and pivotal roles in cancer metabolism.
The GLUT1–4 family of transporters enables the uptake of glucose across the plasma membrane into the cytoplasm13. The comparative analysis of GLUT1 (refs. 14[,15) and GLUT2 (ref. 16) expression in HCC and surrounding tissue reveals a correlation of elevated GLUT1 levels with poor prognosis15. Upon entering the cell, glucose undergoes an irreversible conversion to glucose-6-phosphate (G6P), resulting in a significant increase in the levels of G6P in HCC tumors compared with the surrounding liver tissue17. This conversion is facilitated by a group of enzymes referred to as HKs. HK4 is the HK isoform normally expressed in hepatocytes18, but HK2 is often upregulated in HCC and is closely linked to the clinical stage of the disease and the prognosis of patients19,20. The enhanced ability of HK2 to boost aerobic glycolysis, compared with other HK isoforms, can be attributed to its binding the voltage-dependent anion-selective channel protein-1 located in the mitochondrial membrane. This interaction increases access to mitochondrially generated ATP21,22. Thus, hepatic HK2 deletion inhibits tumorigenesis and increases cell death both in vitro and in vivo19,21. The last enzyme in glycolysis is PK, which transfers phosphate from phosphoenolpyruvate (PEP) to ADP to yield ATP and pyruvate. The M2 isoform of PK (PKM2) is observed mainly in rapidly proliferating cells23 and has been correlated with tumor formation in HCC1,24,25. The conversion of pyruvate to lactate is facilitated by lactate dehydrogenase (LDH), and elevated levels of LDH26 in the bloodstream have been linked to poor progression-free survival in patients with HCC27. Indeed, high levels of LDH and secreted lactate are common features of HCC.
Transcriptomic and metabolomic studies in HCC have shown depletion of major metabolites such as glucose, glycerol 3- and 2-phosphate and malate, alongside an increase in glycolytic activity over mitochondrial oxidative phosphorylation28. The upregulation of glycolytic enzymes is often driven by aberrant activation of the Wnt/β-catenin and PI3K/Akt/mTOR pathways, which are pivotal in HCC’s altered glucose metabolism29,30. Additionally, the expression of the glucose-responsive transcription factor carbohydrate responsive element-binding protein (ChREBP) is elevated in HCC and correlates with tumor aggressiveness. ChREBP is a key regulator of glycolysis, the pentose phosphate pathway and lipogenic genes31. Therefore, given its crucial role in hepatic energy metabolism, ChREBP may represent a promising target for therapeutic intervention in HCC.
Targeting glycolysis has therapeutic potential32,33, as evidenced by strategies that disrupt HK2 function34 or inhibit other glycolytic enzymes35,36,37,38. The hypoxic tumor microenvironment and resulting induction of expression of GLUT1, LDH and hypoxia-inducible factor-1 alpha, further exemplifies metabolic adaptation in HCC6,14,39. In conclusion, HCC displays reprogrammed glycolysis, which is essential for tumorigenesis.
Activation of the pentose phosphate pathway
The pentose phosphate pathway (PPP) is crucial for cancer cell proliferation. Oncogenic metabolic reprogramming encompasses an increase of metabolic flow through the PPP to enhance the supply of nucleotides and reducing equivalents40. In particular, increased PPP diverts G6P to produce ribose 5-phosphate (R5P)41 and NADPH. R5P is essential for nucleotide synthesis, while NADPH plays a key role in lipid synthesis and acts as a major antioxidant by managing cellular levels of reactive oxygen species (ROS).
G6P dehydrogenase (G6PD), the rate-limiting enzyme of oxidative PPP, catalyzes the conversion of G6P to 6-phosphogluconolactone42. In HCC, G6PD is often upregulated43,44, enhancing aspects of malignancy such as apoptosis resistance, migration, invasion and the epithelial–mesenchymal transition45,46,47. The underlying mechanism by which G6PD expression and activity are controlled in HCC remains poorly understood. In clinical settings, the upregulation of G6PD expression has been observed to promote chemoresistance in HCC cells. This can be attributed to the activation of the ID1–WNT–β-catenin–MYC signaling pathway45. In addition, mTORC1 enhances the flux of substances through PPP by facilitating glycolysis, which provides substrates for PPP, and by elevating the levels of G6PD and R5P isomerase A48. The action of nuclear factor-erythroid 2-related factor 2 (NRF2) is also crucial for the expression of G6PD. Mice lacking NRF2 demonstrate resistance to HCC induced by diethylnitrosamine as a result of decreased production of enzymes associated with PPP49.
Transketolase (TKT) is also a critical PPP enzyme. TKT’s activity is regulated by competition between the transcription factors NRF2 and BACH1, with NRF2 enhancing TKT expression to convert glucose derivatives into glutathione, thereby protecting cells from ROS-induced damage50. Elevated TKT levels in HCC are associated with increased tumor aggressiveness51 and poor response to treatments with the TKI sorafenib50.
The intricate regulation of PPP by factors such as NRF2 and the WNT and mTORC1 pathways suggests that these factors could be targeted to improve patient outcomes in HCC.
Dependence on specific amino acids
Glutamine metabolism
The nonessential amino acid glutamine plays a pivotal role in HCC metabolism, serving as a carbon source for anaplerosis and as a nitrogen source for nucleotide and amino acid synthesis52,53. It also contributes significantly to other biosynthetic processes, including anti-ROS glutathione/NADPH production and lipid synthesis54. Notably, altered glutamine metabolism involves the upregulation of specific transporters and enzymes such as sodium-coupled neutral amino acid transporters (SLC38A1 and SLC1A5/ASCT2) and of glutaminase 1 and 2 (GLS1 and 2), enhancing glutamine uptake and its conversion to alpha-ketoglutarate (α-KG)55,56, respectively (Fig. 2a). GLS1 upregulation is particularly pronounced compared with GLS2, which is the isoform typically found in hepatocytes. The glutaminases catalyze the first step in the conversion of glutamine to α-KG, a two-step deamination process. This change in glutaminolysis, often regulated by MYC57, correlates with disease progression and has implications for patient prognosis58.
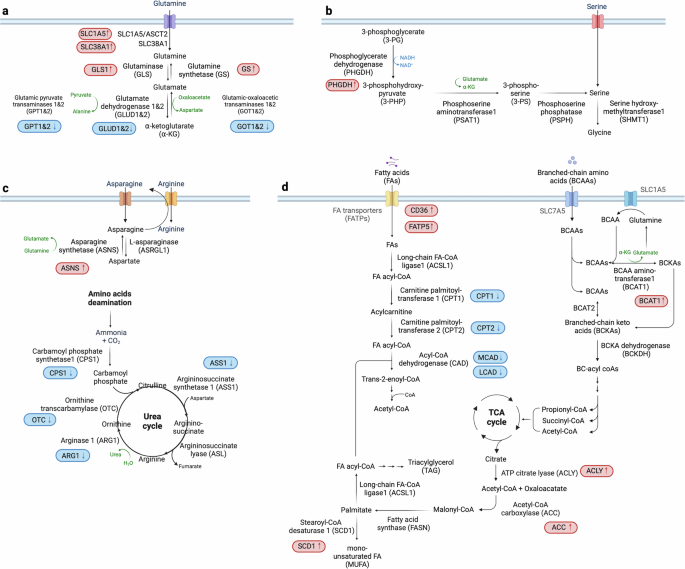
a–d, The critical dependencies and alterations in amino acid (a–c) and FA metabolism within HCC cells (d). Glutamine is pivotal for nucleotide and amino acid production and antioxidant synthesis. In HCC, enhanced glutamine uptake is facilitated by upregulated transporters such as SLC38A1 and SLC1A5, while the conversion to α-ketoglutarate is suppressed due to downregulation of glutamate dehydrogenase 1 and 2 (GLUD1 and 2), promoting glutaminolysis. Elevated levels of GS are associated with increased cell proliferation, highlighting its complex role in cancer progression (a). Serine is essential for nucleotide synthesis and maintaining redox balance, derived from external sources and metabolic pathways from glucose or glutamine. Increased activity of PHGDH boosts de novo serine synthesis in HCC (b). The urea cycle and arginine metabolism involve increased ammonia production and disruptions in metabolism leading to elevated nitrogen supply for biosynthesis. The downregulation of enzymes such as CPS1, ASS1, ARG1 and OTC results in abnormal ammonia metabolism, while dysregulated arginine metabolism promotes oncogenic activities (c). BCAAs undergo conversion to BCKAs, crucial for nucleotide biosynthesis and energy production via the TCA cycle and linked to FAO pathways. Fatty acid metabolism is characterized by increased de novo synthesis and altered FAO. Key enzymes such as ACLY, ACC, FASN, SCD1 and CPT1 are dysregulated, contributing to the metabolic reprogramming that supports tumor growth and progression (d). Proteins with increased levels in HCC are marked in red, while those decreased are shown in blue, providing a clear visual differentiation of metabolic changes in HCC.
Glutamine synthetase (GS), regulated by the Wnt/β-catenin pathway, is another important enzyme in glutamine metabolism. Its level is associated with increased cell proliferation and varied prognostic outcomes59,60. Indeed, emerging studies suggest a complex role for GS in HCC61, potentially linked to cellular differentiation and tumor behavior62,63. Furthermore, the dysregulation of glutamine metabolism impacts several downstream signaling pathways, notably mTORC1 (refs. 64,65) and mTORC2–AKT–C-MYC66, affecting cellular bioenergetics and contributing to the oncogenic processes in HCC. The hepatocyte growth factor axis also influences these metabolic pathways, underscoring the intricate interplay between growth factor signaling and metabolic reprogramming in cancer progression67.
Serine metabolism
Serine is an essential amino acid crucial for nucleotide synthesis and redox homeostasis. It is predominantly synthesized de novo from glucose or glutamine metabolites, but can also be obtained from extracellular sources. Notably, studies have shown that serine levels are significantly elevated in the serum of patients with HCC compared with healthy individuals68, suggesting altered serine metabolism in the tumor environment.
At the molecular level, the transcription factor NRF2 plays a vital role in HCC by regulating intracellular ROS and enhancing the activity of phosphoglycerate dehydrogenase (PHGDH) (Fig. 2b). PHGDH catalyzes the first step in serine biosynthesis, and its upregulation leads to increased serine synthesis69, underscoring the importance of NRF2 in maintaining cellular redox balance and promoting tumor growth. Moreover, hyperactivation of PHGDH and subsequent serine accumulation are associated with poor prognosis in patients with HCC70. PHGDH has been implicated as a driver of sorafenib resistance in HCC, and treatment with the PHGDH inhibitor NCT-503 has been shown to synergize with sorafenib, effectively inhibiting HCC growth in vivo71. Studies have also indicated that serine is not merely a metabolic byproduct, but a critical substrate in HCC pathophysiology. A deficiency in serine impedes HCC cell growth, highlighting serine metabolism as a vulnerability in cancer cells69.
Urea cycle, arginine and asparagine metabolism
The urea cycle is fundamental to liver function, converting harmful ammonia, a byproduct of amino acid and protein catabolism, into urea for excretion. Key enzymes in this process include carbamoyl phosphate synthetase 1 (CPS1), argininosuccinate synthetase 1 (ASS1), argininosuccinate lyase (ASL), arginase 1 (ARG1) and ornithine transcarbamylase (OTC)72,73. In HCC, disruptions in this cycle contribute to abnormal ammonia metabolism and an increased availability of nitrogen for nucleotide synthesis, supporting rapid tumor growth (Fig. 2c).
HCC is characterized by epigenetic downregulation of CPS1 and ASS1 through hypermethylation74, impacting cell proliferation and apoptosis. In particular, ASS1 suppression leads to arginine autotrophy74,75 that enhances tumor cell migration, invasion and metastasis76. ARG1 catalyzes the breakdown of arginine into ornithine and urea. Ornithine then undergoes additional metabolism to form polyamines, which mediate numerous cellular processes75,77. ARG1 is the predominant isoform in the hepatic urea cycle, whereas ARG2 is expressed mainly in extrahepatic tissues78. Arginine, an amino acid that is considered conditionally essential, plays a role in various biological processes and has been associated with tumorigenesis79. Arginine is taken up into the cell mainly via SLC7A1, and HCC shows a high amount of intracellular arginine80,81. The arginine mediates further metabolic reprogramming by binding directly to the transcription co-factor RBM39 (ref. 82). It is noteworthy that the synthesis of asparagine exhibits a strong correlation with arginine uptake in HCC82, probably due to the use of asparagine as an anti-solute in arginine import.
The synthesis of asparagine occurs through an ATP-dependent process catalyzed by asparagine synthetase (ASNS). Glutamic acid is utilized as the nitrogen source in this process83. The expression of ASNS is increased in HCC tumors and is associated with the tumor stage and prognosis. Suppression of ASNS hinders the proliferation, migration and development of HCC cells, indicating its potential as a target for therapeutic intervention84. Furthermore, ARG1 expression is substantially decreased in HCC. The combined effects of increased asparagine-dependent arginine uptake and decreased ARG1-dependent arginine conversion to polyamines account for the elevated levels of arginine in liver cancer cells. This metabolic rewiring further mediates the development of oncogenic metabolism and tumor progression, highlighting the intricate involvement of arginine in the pathophysiology of liver cancer82. Lastly, OTC, which catalyzes the conversion of ornithine and carbamoyl phosphate into citrulline72, is crucial for ammonia detoxification. Deficiencies in OTC can lead to ammonia accumulation, a risk factor for HCC development85,86.
These metabolic shifts in the urea cycle and associated amino acid metabolism not only provide a deeper understanding of HCC pathogenesis, but also reveal potential targets for therapeutic intervention. Addressing the dysregulation of enzymes such as CPS1, ASS1, ARG1 and OTC could offer new strategies to curb HCC progression by manipulating the tumor’s metabolic dependencies.
BCAA metabolism
Branched-chain amino acids (BCAAs), which include leucine, isoleucine and valine, are essential amino acids distinguished by their nonlinear structure. In contrast to other amino acids, BCAAs are not endogenously produced by the human body and need to be obtained via the diet. These amino acids have a vital function in maintaining cell growth by serving as a nitrogen and/or carbon source87.
The isoenzymes BCAA aminotransferase 1 and 2 (BCAT1 and 2), which are located in the cytoplasm and mitochondria, respectively, convert BCAAs into the corresponding branched-chain keto acids (BCKAs) (Fig. 2d). BCKAs subsequently undergo oxidative decarboxylation catalyzed by the BCKA dehydrogenase (BCKDH) complex, resulting in the formation of acyl-coenzyme-A (acyl-CoA)88. The next steps of BCAA breakdown are similar to those of fatty acid oxidation (FAO), resulting in the formation of end products that can enter the TCA cycle.
The catabolism of isoleucine leads to the production of acetyl-CoA and propionyl-CoA, leucine produces acetyl-CoA and valine yields propionyl-CoA. These products are utilized by the TCA cycle, highlighting the role of BCAA catabolism in energy metabolism and cellular growth. This process is linked to cancer development, with BCAAs having a dual role of either promoting or inhibiting tumor growth depending on the genetic and tissue context89,90,91. Recent studies have shown that BCAAs act as signaling molecules that reflect the body’s nutritional state. As such, they can influence metabolic pathways related to cancer progression92 and increase the risk of HCC93. Also, enhanced breakdown of BCAAs in HCC and the redirection of their metabolic products toward nucleotide synthesis support rapid cell division and tumor growth. Furthermore, the expression of BCAT1 is elevated in HCC, correlating with increased tumor cell migration and invasion94. Conversely, long-term administration of BCAAs has been observed to improve the nutritional status and longevity of patients with liver cirrhosis, indicating a protective role under certain conditions95,96,97. BCAAs have demonstrated the ability to inhibit HCC caused by diethylnitrosamine in obese mice98 and rats99. Nevertheless, a decrease in the activity of BCAA catabolic enzymes results in elevated levels of BCAA and heightened activation of mTORC1, hence facilitating the growth of tumors100. Despite these insights, the complete mechanism by which BCAAs influence HCC development remains poorly understood and is a subject of ongoing research.
Changes in FA metabolism
FA uptake and synthesis
The liver has an important role in mediating systemic lipid homeostasis, which includes the uptake, synthesis, storage, release and breakdown of fatty acids (FAs). FAs come from de novo synthesis within the liver or from the breakdown of fats stored in adipocytes during fasting or insulin resistance. Hepatocytes utilize fatty acid transporters (FATPs) and FA-binding proteins to facilitate FA uptake from the bloodstream. In the context of HCC, the metabolic processes of FA uptake and synthesis are significantly altered (Fig. 2d). Notably, the expression of FA transporters such as CD36 and FATP5 is upregulated in HCC, underscoring the importance of extracellular FA sources in cancer progression101,102. While normal cells predominantly rely on circulating exogenous FAs, HCC cells exhibit elevated de novo FA synthesis alongside uptake. This affects the process of membrane formation and pathways involved in signaling103.
The synthesis of FAs begins with the conversion of citrate to acetyl-CoA, catalyzed by ATP citrate lyase (ACLY). Subsequent reactions involving acetyl-CoA carboxylase (ACC) and fatty acid synthase (FASN) lead to the production of FAs. Stearoyl-CoA desaturase 1 (SCD1) then desaturates these FAs to produce monounsaturated FAs (MUFAs), crucial for triacylglycerol (TAG) synthesis104. The overexpression of enzymes such as ACLY105, ACC105,106, FASN105,107 and SCD1 (ref. 105) is commonly observed in HCC, enhancing lipid synthesis pathways and contributing to tumor growth.
AMP-activated protein kinase (AMPK) phosphorylates and thereby inhibits ACC, thus exerting an influence on hepatic de novo FA synthesis and potentially affecting carcinogenesis108,109. Consistent with this notion, a novel liver-specific ACC inhibitor, ND-654, mimics the effects of ACC phosphorylation, inhibiting hepatic de novo FA synthesis and HCC development109. Similarly, SCD1 which is involved in the synthesis of MUFAs, is associated with tumor proliferation in xenograft animal models and the survival rates of patients27. Although FASN does not directly transform hepatocytes into cancer cells, its role is crucial in the progression of hepatic steatosis and tumor development, particularly influenced by AKT signaling107. A preclinical study suggests that TVB3664, a novel FASN inhibitor, significantly ameliorates the fatty liver phenotype, although TVB3664 monotherapy shows moderate efficacy in metabolic dysfunction-associated steatohepatitis (MASH)-related murine HCCs110.
The transcription factor sterol regulatory element-binding protein-1 (SREBP-1) orchestrates the expression of FA synthesis genes, significantly impacting cellular proliferation111, migration111 and survival in HCC112,113,114. Dysregulation of SREBP-1, along with aberrant mTORC1 and mTORC2 signaling79,115,116,117,118, further complicates FA metabolic reprogramming in HCC. For instance, the deletion of hepatic tuberous sclerosis 1 (TSC1) a suppressor of mTORC1 signaling, has been demonstrated to stimulate mTORC1 and trigger the formation of spontaneous HCC with slight FA accumulation119. Notably, another study revealed similar cases of spontaneous HCC formation in mice lacking TSC1. However, in this particular case, the observed characteristics were ascribed to defects in the maturation of SREBP-1 and de novo FA synthesis, rather than direct activation of mTORC1 (refs. 120,121). The aforementioned results highlight the relationship between mTORC1 signaling, involving Akt, and lipid metabolism within the framework of hepatocarcinogenesis. It is worth noting that the activation of mTORC1 alone does not adequately stimulate hepatic SREBP-1. However, when rictor, a component of mTORC2, is specifically knocked out in the liver, there is a decrease in Akt Ser473 phosphorylation and a reduction in SREBP-1 activity, resulting in defective lipogenesis and hypolipidemia118. In addition, IGF-induced phosphorylation of Akt Ser473 leads to Akt-mediated phosphorylation of cytosolic phosphoenolpyruvate carboxykinase 1 (PCK1), the rate-limiting enzyme in gluconeogenesis. Phosphorylated PCK1 translocates to the endoplasmic reticulum, activating SREBP and enhancing the transcription of lipogenic genes by disrupting the interaction between INSIG proteins and SREBP cleavage-activating protein122. Thus, mTORC2 mediates hepatic lipid metabolism via insulin-induced Akt signaling. This underscores the complex interplay between the mTORC1 and mTORC2 pathways in the context of HCC progression. Furthermore, in animals lacking both tumor suppressors PTEN and TSC1, mTORC2 has been linked to the development of HCC, partly by activating SREBP-1 and facilitating FA production. This study provides a comprehensive understanding of the function of mTORC2 in HCC through its regulation of lipid metabolism, hence offering insights into prospective treatment strategies123.
FA breakdown
The liver possesses the capacity to metabolize FAs via both the mitochondrial and peroxisomal β-oxidation pathways (FAO). Carnitine palmitoyltransferase 1 (CPT1)124, essential for transporting FAs into mitochondria for oxidation, shows decreased activity in HCC contexts125,126,127 (Fig. 2d). This dysfunction contributes to the oncogenic potential of FA metabolism in HCC55. A thorough examination of metabolic profiles in obesity-induced HCC mouse models revealed a notable accumulation of long-chain acylcarnitine species, which is attributed to reduced conversion of acylcarnitine to acyl-CoA, probably due to reduced expression of CPT2. The downregulation of CPT2 leads to suppression of FAO, contributing to the steatotic changes commonly seen in HCC. Moreover, CPT2-mediated lipid metabolic reprogramming not only facilitates the adaptation of HCC cells to a lipid-rich microenvironment, but also drives liver tumor progression128. Previous studies have further demonstrated reduced CPT2 expression in patients with MASH-related HCC128, alongside elevated acylcarnitine levels in HCC mouse models and patients129. These findings suggest a potential metabolic shift that may contribute to the progression of obesity and MASH-related HCC. In addition, it is worth noting that medium-chain acyl-CoA dehydrogenase (MCAD) and long-chain CAD (LCAD), dehydrogenases involved in the initiation of FAO, display reduced expression in HCC tumors. MCAD has a role in the progress of HCC by interfering with the pathways involved in FAO, while the reduction in LCAD facilitates the progression of HCC by causing an accumulation of unsaturated FAs within cells113,130. The aforementioned findings underscore the relationship between the dysregulation of FAO and the development of HCC.
Combining multiple metabolic reprogramming events
Metabolic reprogramming involves alterations across multiple biochemical pathways. One common aspect is a disturbance in acetyl-CoA metabolism, which is central to various physiological processes such as the TCA cycle, lipid synthesis and protein acetylation131. In addition to via breakdown of FAO and BCAA in cells, acetyl-CoA is also generated through the oxidative decarboxylation of pyruvate obtained from glycolysis132. In HCC, there is a noted reduction in acetyl-CoA production133 due to transcriptional downregulation of acetyl-CoA synthesis pathways, including FAO128, BCAA catabolism100 and pyruvate catabolism (Fig. 3a). This reduction impacts not only energy metabolism, but also the acetylation of histone and non-histone proteins, influencing gene expression and cellular differentiation. The downregulation of acetyl-CoA levels in HCC is mediated by the two transcription factors TEAD2 and E2A. These transcription factors, via downregulation of acetyl-CoA biosynthesis genes, drive proliferation and dedifferentiation in HCC, ultimately leading to poor patient survival133.
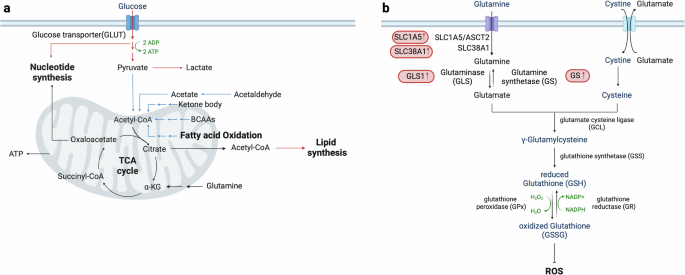
The intricate interactions between various metabolic pathways that influence acetyl-CoA metabolism and redox homeostasis in HCC cells. a, Acetyl-CoA production is significantly reduced in HCC due to the downregulation of its biosynthetic pathways. This reduction impacts both energy metabolism and protein acetylation, subsequently affecting gene expression and cellular differentiation. b, HCC cells manage oxidative stress by enhancing antioxidant defenses, primarily through the glutathione–glutamine–glutamate pathway. Despite elevated levels of ROS, these adaptive mechanisms are essential for maintaining redox homeostasis, which is crucial for cancer cell viability, progression and recurrence. Upregulation of GLS1 in HCC increases glutamate production, thereby boosting antioxidant capacity and promoting characteristics associated with cancer stemness. Proteins and pathways that are upregulated in HCC are marked in red, while those that are downregulated are indicated in blue, providing a clear visual representation of the metabolic changes in HCC.
Oxidative stress is due to an imbalance of ROS production and antioxidant defenses134,135,136. Despite increased ROS, HCC cells maintain redox homeostasis through robust antioxidant mechanisms135, notably involving the glutathione–glutamine–glutamate pathway. In HCC, imported glutamine is converted to glutamate, which is a precursor for the synthesis of glutathione (Fig. 3b). Glutathione, a potent antioxidant, protects cancer cells from oxidative harm, thereby facilitating cellular viability and proliferation. The upregulation of GLS1 in HCC cells enhances glutamate production, which, in turn, increases antioxidant capacity and gene expression associated with stemness137. The maintenance of redox homeostasis is important for the progression and recurrence of HCC138, since oxidative damage enhances susceptibility to the illness68. Furthermore, HCC exhibits indications of DNA and lipid oxidative damage139, while patients with MASH-related HCC demonstrate decreased serum antioxidative function compared with patients with metabolic dysfunction-associated fatty liver disease140. These results collectively highlight the complex relationship between metabolic reprogramming and hepatocarcinogenesis, emphasizing the diverse functions of metabolism in promoting the development and progression of HCC.
Conclusion
Oncogenic metabolic reprogramming is a dynamic and complex process influenced by numerous inputs responding to various cellular conditions. This reprogramming results in highly adaptable cellular states allowing survival and proliferation under diverse and challenging conditions. The interconnected nature of metabolism means that alterations in one pathway can significantly impact others, creating a network of metabolic changes that collectively contribute to cancer progression. Understanding the multilevel control of this reprogramming may reveal metabolic vulnerabilities, opening promising new avenues for therapeutic strategies. Such precision medicine approaches have the potential to enhance the effectiveness of treatments by directly targeting the unique metabolic dependencies of tumor cells.
Responses