New fast ion conductors discovered through the structural characteristic involving isolated anions

Introduction
Nowadays, lithium ion batteries are becoming increasingly popular throughout the fast growing markets of mobile electronic devices, electric vehicles and energy storage technologies1,2. Compared with currently used lithium ion batteries based on organic liquid electrolytes, all-solid-state lithium batteries have caught more and more attention3,4,5,6. In all-solid-state lithium batteries, the design of solid-state electrolytes is crucial7. Up to now, some solid electrolytes have been discovered and well explored8. Li argyrodites9,10, Li10GeP2S12 (LGPS)11,12, NASICON-type LiM2(PO4)3 (M = Ge, Ti, Sn, Hf, Zr)13, garnet-type LixLa3M2O12 (5 ≤ x ≤ 7, M = Nb, Ta, Sb, Zr, Sn)14,15, halide-based Li-M-X (M = metal element, X = F, Cl, Br, I)16,17 and so on exhibit ionic conductivity similar or close to that of liquid electrolytes, but the mystery of the relationship between lattice atomic structure and ionic conductivity remains unresolved. Lots of studies attempt to summarize the characteristics of current fast ion conductors18,19 and try to discover more high-performance solid-state electrolytes20,21. Despite the property of mobile cations in the lattice, the function of the framework consisting of immobile ions offers another perspective for research. Jun et al.22 claimed that corner-sharing connectivity of the oxide crystal structure framework promotes superionic conductivity. Wang et al.18 revealed the structural feature of face-sharing high-coordination sites for fast sodium-ion conductors. Furthermore, similar studies of the characteristic of anions in framework structures have been made. Wang et al.17 believed that a structure with a bcc anion lattice is more likely to show high ionic conductivity, and the closer the anion lattice is to a perfect bcc lattice, the higher the conductivity. Treating all anions as an entirety is beneficial for identifying their spatial connectivity features, but overlooks their specific features in the local environment. In this work, we identified a special type of anions which do not bond with immobile cations in the lattice, but only form weak bonds with moving lithium ions. We refer to them as isolated anions. We found that the presence of isolated anions can lead to the frustration phenomenon and facilitate Li+ ion transport by forming a flat potential energy surface (PES) around themselves to reduce the Li+ migration barriers.
The frustration in superionic conductors, acting as one of the effective migration mechanisms for high conductivity of Li+ ions, might be caused by local distortion or by partial occupation. For example, Stefano et al.23 showed that the disorder distortion of [PS4]3- groups in LiTi2(PS4)3 triggers the frustration phenomenon by offering no energetically stable tetrahedral or octahedral positions for the Li+ ions in the structure. Wang et al. 24 proposed the Density Of Atomistic States (DOAS) to quantitatively characterize the degree of disordering and elucidated how the frustration enhances diffusion. The distribution of DOAS also reflects the similarity of local environments for Li+ ions on the smooth PES. Although the role of frustration phenomenon in ion transport has been recognized, there still exist mysteries such as the features of structures with frustration, the criteria for determining if a frustration appears and the design method for frustrated system.
In this work, we found the frustration phenomenon induced by isolated anions and its direct cause through analyzing the structure and ionic conducting characteristics of Li6PS5Cl, and by thoroughly investigating the ion migration behaviors in a series of structures of the prototype compound Li8SiSe6, we further confirmed that isolated anions can be one of the effective feature environments for introducing frustration in crystal structures. The occurrence and causes of the frustration induced by isolated anions, the definition of isolated anions, as well as how their local symmetries correlate with the appearance of frustration, and how the arrangement and the element type of anions affect the ionic transport are discussed. Using isolated anions as the structural feature, we screened compounds containing this feature from the crystal structure database, and further AIMD studies confirm the fast ion migration behavior in these compounds, proving the function of isolated anions in creating high ionic conductivity in these structures.
Results
The frustration induced by isolated anions
The role of isolated anions in the frustration effect is found inspired by the ion transport characteristics of lithium argyrodite Li6PS5X (X = Cl, Br)9 system, in which the anion lattice is close to the fcc configuration and deviates far from the bcc one but shows ionic conductivity as high as 1.1 × 10–3 S/cm25. Further looking into the lattice, two different types of anions with distinct chemical environments — isolated anions and bonded anions, can be found. As shown in Fig. 1a, isolated anions (Siso, Cliso) are considered as the S or Cl anions those do not form chemical bonds with P atoms but only bond with Li ions, and they occupy Wyckoff positions 4 d and 4a. On the other hand, bonded anions (Sbond) refer to the S anions that form PS4 tetrahedra by bonding with P atoms, and they occupy Wyckoff position 16e.

a Crystal structure of Li6PS5Cl. b Li+ diffusion pathways in Li6PS5Cl calculated through AIMD simulation, the areas within the blue surfaces are positions with high probability density of Li+ ions. c The local environment of Siso (the inset in the orange box), Cliso (the inset in the green box) and Sbond (the inset in the blue box) and the partial electronic density of states (PDOS) of Siso, Cliso and Sbond in Li6PS5Cl. d A schematic diagram of energy landscape for Li+ migration in normal structure and structure with frustration.
We found that the presence of frustration phenomenon in Li6PS5Cl is induced by isolated anions as aforementioned. The different local structures of Siso, Sbond and Cliso, as well as their various PDOS of Li6PS5Cl in Fig. 1c demonstrate the inconsistency of their chemical environments. In previous studies, the transport of Li+ in argyrodites was divided into three types of events, named as doublet jump, intra-cage hoppings and inter-cage hoppings26. According to the Li+ diffusion pathways in Li6PS5Cl revealed by AIMD simulations shown as Fig. 1b, the isolated anions Siso play a role as the center of each cage, implying that on the cage centered around Siso there exists a smooth PES for Li+ ions, which is a characteristic indicator for frustration phenomenon in fast ion conductors. In general, the frustrated system has a large number of degenerated states with similar energies. The exceptionally smooth energy landscape, comparing with normal structures with specific stable sites for Li+ ions (Fig. 1d), ensures that the system can achieve lower energy barrier transitions between degenerated states. The distance of atomic descriptors between positions in the structure can tell the degree of the similarity in the local chemical environments and local configuration energy like DOAS. By computing the Euclidean distances of Atom-Centered Symmetry Functions (ACSFs) descriptor27 between positions in the lattice of Li6PS5Cl (Supplementary Fig. S1 and Supplementary Note 1), the positions similar to Li+ lattice sites were picked out and the results show that they are all distributed on Li cages surrounding the isolated anions, forming a sphere with many low-energy equivalent sites around the isolated anions, which means that the similar local chemical environment creates a spherical potential energy surface causing Li+ ions to exhibit a frustration arrangement on this sphere, indicating the presence of frustration around Siso. Therefore, through the analysis of Li6PS5Cl, we found that the presence of isolated anions can generate a spherical potential field around it, causing frustration in the crystal structure.
Selection of prototype system Li8SiSe6
There are many problems worth exploring regarding the isolated anions. Just as solid-state electrolytes Li3PS428,29, Na3PS430, Li2B12H12, LiCB11H1231 and Li7La3Zr2O1232,33, most argyrodites show different phases at the high- and low-temperature regions34. Generally, the high-temperature phase has higher symmetry as well as higher ionic conductivity. Also, the doping of halogen elements (Cl, Br) helps to stabilize the high-temperature phase at room temperature35, for example, Li6PS5Cl, but the Cliso or Briso anions do not generate Li cages around them as Siso do. Therefore, the symmetry of the structures, the arrangements and the types of isolated anions are all important factors related to frustration and Li+ transport. In order to study these issues, we need to select a system that can incorporate all of the above variables. To this end, we have chosen a series of Li8SiSe6 structures and the reasons for choosing them as prototype structures are as follows. Firstly, Li8SiSe6 structures contain both isolated Se (Seiso) and bonded Se (Sebond), which is necessary for our research. Secondly, the diverse and abundant phase structures of Li8SiSe6 can be derived from other existing argyrodite materials through element substitution (Table 1), and these phases have varying levels of ion transport capabilities, providing the possibility to construct relationship between various structural variables and ionic conductivity. Thirdly, phases of Li8SiSe6 show relatively high ionic conductivities at 300 K or 400 K compared with other argyrodites, eliminating the necessity for extrapolating room temperature conductivity by high-temperature AIMD simulations, hence the influence of phase transition caused by temperature change can be removed. Finally, in Li8SiSe6, there is only one type of anion element, Se, which reduces the influence of doped halogen element such as Cl or Br, making less variables and making the initial research simpler and more intuitive. Halogen elements can also be introduced to replace part of isolated Se sites for comparative research to clarify the effects of anion types. Overall, six different space group structures of Li8SiSe6 (Li8SiSe6_F(bar{4}) 3 m, Li8SiSe6_Pna21, Li8SiSe6_Pmn21, Li8SiSe6_P63cm, Li8SiSe6_Cc and Li8SiSe6_hcp) as well as Li7SiSe5Cl with F(bar{4}) 3 m space group were considered in our research system (Table 1).
Definition and characteristics of isolated anions
Anions those exist independently in structures and do not form local structures such as tetrahedral or octahedral coordinating with immobile non-lithium cations, are defined as isolated anions in our study. As shown in Fig. 2a, the Se2− anions are classified into Sebond and Seiso according to whether they appears at the corner of [SiSe4]4− tetrahedra or as the isolated one only surrounded by Li+ ions. Besides the different bonding characteristics, these two types of Se anions also exhibit clear differences in their electronic structures and vacancy formation energy. The PDOS and vacancy formation energy for Sebond and Seiso in Li8SiSe6 argyrodites are shown in Fig. 2b, c and Supplementary Fig. S2. In Li8SiSe6_F(bar{4}) 3 m, the PDOS of Seiso overlaps with part of Li’s PDOS and no superimposing PDOS between Seiso and Si, indicating bonds with only Li, and the PDOS of Seiso1 and Seiso2 are distributed in different energy range due to their distinct bonding numbers to Li as shown in Fig. 2a. However, the PDOS of Sebond overlaps with PDOS of both Si and Li, implying bonds with both Si and Li. The vacancy formation energy is the energy required to create a vacant defect in the crystal lattice, indicating the difficulty level of vacancy formation, and represents the bonding characteristics and the stability of the atom at that location. The higher vacancy formation energy of an atom means stronger interactions with neighboring atoms in the structures36,37. As shown in Fig. 2c, the vacancy formation energy of Seiso is larger than that of Sebond which illustrates that Seiso is more stable in structures. In this structure, the number of interacting Li-Seiso neighboring atom pairs is much greater than the number of interacting Si-Sebond and Li-Sebond neighboring atom pairs, resulting to Seiso being more stable and showing higher vacancy formation energy. This feature can be effectively used to distinguish between bonded and isolated anions in the structure. The values also partially explain why Sebond is easy to leave its lattice position and rotate around the central Si atom, while Seiso prefers stable at its lattice site. The unique local environment of isolated anions makes them show strong influence on Li+ ion transport. So next, after evaluating the ionic conductivity in each structure, we will figure out potential influencing factors one by one and study how the isolated anions affect Li+ ion transport and their correlation with frustration mechanism.
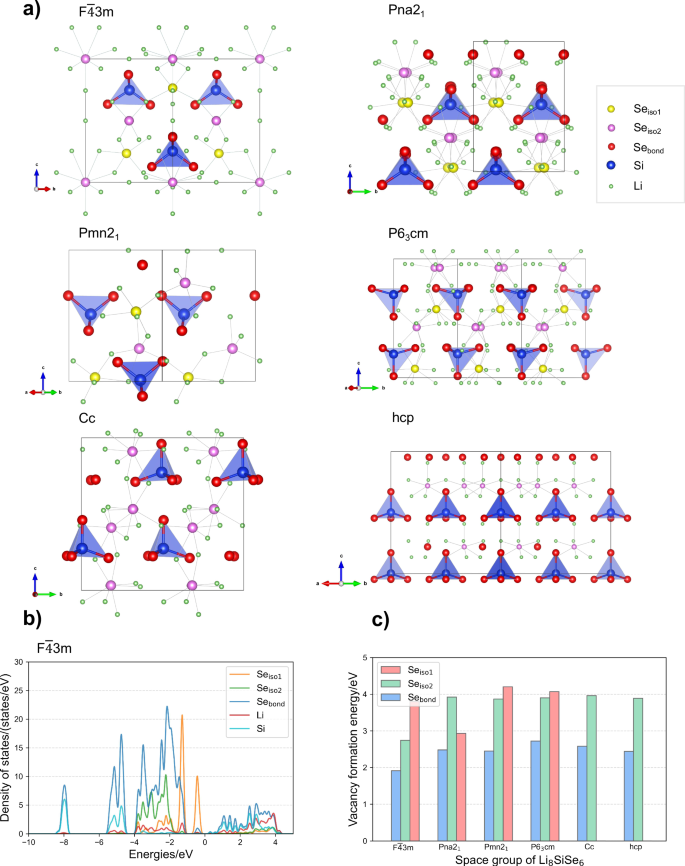
a The structures of Li8SiSe6. b PDOS in Li8SiSe6_F(bar{4}) 3m. c Vacancy formation energy of Seiso and Sebond in Li8SiSe6 structures. In Li8SiSe6_F(bar{4})3m, Li8SiSe6_Pna21, Li8SiSe6_Pmn21, and Li8SiSe6_P63cm, Seiso can be divided into Seiso1 and Seiso2 due to different Wyckoff positions. The calculation process of vacancy formation energy is described in Supplementary Note 2.
The thermodynamic stability and Li+ ion transport properties
The thermodynamic stability for each Li8SiSe6 structure is evaluated by calculating the convex hull energy (Ehull) as shown in Table 1. Ehull refers to the calculated enthalpy of formation for the material by determining its decomposition products through convex hull analysis38. The Ehull value is a non-negative number, with a higher value indicating poorer thermodynamic stability of the structure and a greater possibility of decomposing into other stable phases. According to the values listed in Table 1, Li8SiSe6_hcp is thermodynamically stable, while other Li-Si-Se phases studied in this work are thermodynamically metastable. Nevertheless, in this study, the Li8SiSe6 system was selected for investigating how various structural variables affect the migration of Li+ ions in the presence of isolated anions, and it is not the fast ion conductor candidates we screened out. In the later section where promising fast ion conductor materials are screened through the structural characteristic involving isolated anions, we applied the stringent thermodynamic stability criteria, with Ehull less than 15 meV/atom, to ensure the possibility of synthesis and existence of the recommended materials39. Then, the Li+ ion diffusivity and ionic conductivity are evaluated for each Li8SiSe6 structure by fitting the MSD curves statistically extracted from AIMD simulations at 300 K or 400 K (Table 1 and Supplementary Fig. S3). All six structures exhibit a cage-like transport mechanism surrounding the Seiso (Supplementary Fig. S4). Li7SiSe5Cl’s MSDs, diffusivity and conductivity at 300 K are also obtained (Supplementary Fig. S5, Table 1). As Table 1 shows, argyrodites containing Se element can already achieve very high Li+ conductivity without the existence of halogen elements. For Li8SiSe6 in the space group of F(bar{4}) 3m, Pmn21 and P63cm, ion migration events can even be observed at 300 K. Given the close MSD values among the structures Li8SiSe6_F(bar{4}) 3m, Li8SiSe6_Pmn21 and Li8SiSe6_P63cm, we further calculated the MSD of these phases at a higher temperature of 1000 K, which triggers more migration events, thereby amplifying the differences of the MSD values among them (Supplementary Fig. S3e). However, both Li8SiSe6_hcp and Li8SiSe6_Cc structures exhibit no Li+ motion at 300 K, and the MSD of Li+ ions in Li8SiSe6_Pna21 is too small to estimate the diffusivity and ionic conductivity, indicating their lower Li+ ion migration ability compared with F(bar{4}) 3m, Pmn21 and P63cm phases. To distinguish among the structures Li8SiSe6_Pna21, Li8SiSe6_Cc and Li8SiSe6_hcp, we performed AIMD simulations at 400 K for them, and the kinetic properties can be obtained for Li8SiSe6_Pna21 and Li8SiSe6_Cc, while Li8SiSe6_hcp still shows no hopping events at 400 K. Therefore, the ion transport ability for various structures of Li8SiSe6 is ranked as: Li8SiSe6_F(bar{4}) 3 m > Li8SiSe6_Pmn21 > Li8SiSe6_P63cm > Li8SiSe6_Pna21 > Li8SiSe6_Cc > Li8SiSe6_hcp. Additionally, we carried out the AIMD simulation for Li8SiSe6_F(bar{4})3m with fixing the position of all Se atoms located in (SiSe4)4- tetrahedra at 300 K and obtained the MSDs by element to study the possible impact of the rotation of (SiSe4)4- tetrahedra on the Li+ ion migration in Li8SiSe6. In this simulation, all bonded anions are fixed at their initial lattice sites, preventing anion groups from rotating during the simulation and excluding the influence of the rotation of (SiSe4)4- tetrahedra on Li+ ion migration. Supplementary Fig. S6 shows the MSD obtained from the simulation with fixed Sebond anions, and the result with Sebond anions in free motion are also shown in the figure for comparison. It can be seen that the MSD of Li is very similar in both cases, indicating that in this structure, the rotation of bonded anion groups is not closely related to Li+ ion transport (Supplementary Note 3). Further looking into the structures of these Li-Si-Se structures, we found that comparing the spatial positions of cages in Li8SiSe6_F(bar{4}) 3 m and Li7SiSe5Cl, the neighboring cages in Li8SiSe6_F(bar{4}) 3m share a common face, while the cages in Li7SiSe5Cl are far apart resulting in distinguishable intra- and inter-cage structures (Supplementary Fig. S7). In addition, the face-sharing cages also exist in Li8SiSe6_Pna21, Li8SiSe6_Pmn21, Li8SiSe6_P63cm and Li8SiSe6_Cc, but Li8SiSe6_hcp shows a similar case to Li7SiSe5Cl in which cages are apart from each other. We can deduce that different local environments and spatial arrangements of isolated anions have an impact on the formation of cages as well as the ion migration. Therefore, we will systematically discuss the influence of various factors, such as symmetry, arrangement, and elemental type, on the ion migration properties in detail.
The effect of isolated anions’ local structures’ symmetry
The local structural features for Seiso anions in each compound are analyzed to build the relation to the kinetic properties. For a specific Seiso anion, the point group of the adjacent [SiSe4]4− tetrahedra and other Seiso is adopted to describe its local symmetry. The symmetry of local environment can represent the symmetry of Li cages’ PES around Seiso. The intuitive understanding is that a highly symmetric local environment will lead to the PES around Seiso having high symmetry, which is more conducive to the formation of the cage-like pathway for lithium ions.
For the five structures with face-sharing Li+ migration cages as mentioned above, the inter-cage transport can be accomplished through these sharing faces, and thus they are a suitable set of systems for studying the influence of local symmetry on intra-cage transport. The point group of local environments for Seiso in these five structures are determined and shown in Fig. 3a and Supplementary Fig. S8. Three types of point group environment are found, including Td, Cs, and C1. It is found that structures with higher local symmetry also exhibit higher ionic conductivity, which occurs through the hopping of Li+ ions within the cage surfaces, known as intra-cage pathways around Seiso. By controlling synthesis conditions or substitution, the lattice space group can be adjusted by forming various phases and the symmetry of local structures can be controlled to improve ion transport performance in ion conductors40. However, for the remaining structure, Li8SiSe6_hcp, the point group of local environment for Seiso is C3. The high symmetry maintains the high degree of frustration in the region of cage surfaces around Seiso, but the distance between cages restricts the ion migration within the entire crystal framework. As a result, the hcp phase shows relatively low ion conductivity, thus we will explore the conditions for reducing the energy barrier of inter-cage transport from the perspective of isolated anion arrangements.
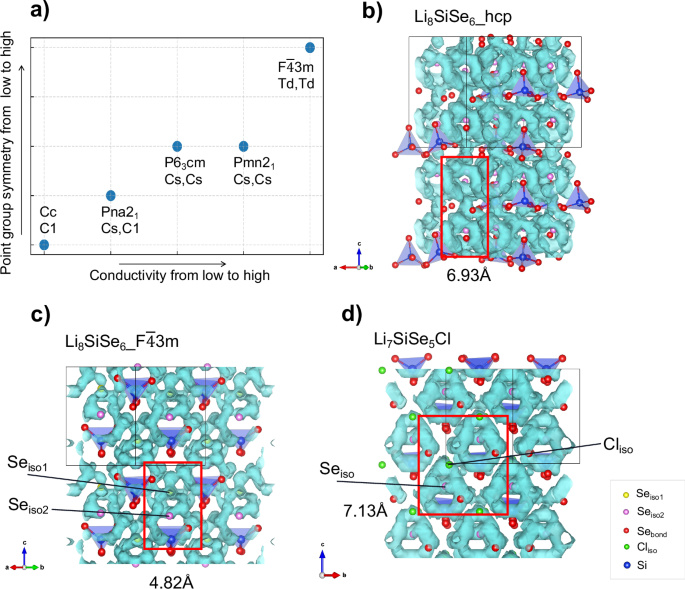
a A schematic diagram of the relationship between Seiso local structure’s point group symmetry and Li+ conductivity. b–d Li+ diffusion pathways obtained from AIMD simulations in Li8SiSe6_hcp, Li8SiSe6_F(bar{4})3m and Li7SiSe5Cl. The values are distances between adjacent Seiso in red boxes. The Li+ ions in (b–d) are hidden for better observation of the pathways.
The effect of the arrangement of isolated anions
Since the isolated anions act as the centers of the cage pathways, their arrangement will determine the position of the cages and affect the ion transfer among these cages. When the isolated anions are closely packed in a structure, coplanar transport can be easily formed to connect pathways among different cages, allowing for inter-cage transfer to occur directly, as found in the five structures in Fig. 3a. The distances between neighboring cage centers are 4.82 Å for Li8SiSe6_F(bar{4}) 3m, 4.45 Å for Li8SiSe6_Pna21, 4.49 Å for Li8SiSe6_Pmn21, 4.27 Å for Li8SiSe6_P63cm and 4.15 Å for Li8SiSe6_Cc. While for structure Li8SiSe6_hcp (Fig. 3b), the distance between adjacent Seiso anions is as large as 6.93 Å, where the cages can’t be connected directly, thereby the inter-cage transport becomes a major limiting factor for Li motion and results in the relatively low Li+ ion conductivity. According to the atomic radius of Li and Se’s, the maximum Seiso spacing of 5.22 Å is estimated as the critical distance to achieve coplanar cage transport. On the other hand, when the distance between Seiso and other non-Li ions is too small to permit Li+ pass, the cages can’t form successfully. Therefore, high conductivity can be obtained by constructing coplanar Li ion cages through shortening the distance of adjacent isolated anions to below critical distance while maintaining the geometric size of the Li+ migration tunnels.
The effect of isolated anions’ element types
When Li8SiSe6_F(bar{4}) 3 m is compared with Li7SiSe5Cl, it is found that even though the Cl-doped F(bar{4})3m structure has much lower Ehull than the original one (Table 1), no Li+ cage pathways around Cliso can be found and only the intra-cage transport around Seiso is maintained. The disappearance of cages around Cliso sites leads to the increase of the distance between isolated anions from 4.82 Å to 7.13 Å, and the face-sharing inter-cage transport are damaged (Fig. 3d), thus the doped structure Li7SiSe5Cl has a lower Li MSD and conductivity at 300 K than Li8SiSe6_F(bar{4}) 3 m (Table 1 and Supplementary Figs. S3, S5). Ouyang et al.41 explained the phenomenon in Na argyrodite by analyzing the Na-Na intracluster and intercluster distances of every structure. They pointed out that halogen doping can change these distances in structures, and the distance variation is created by the size contrast between the halogen and the sulfur. Here we illustrate this phenomenon and characterize it quantitatively from the perspective of interactions between Li+ and isolated anions.
For the cases with two different types of anionic elements, Li+ ions may move closer to anions with stronger interactions and form cage-like pathways around them, while the probability of lithium ions appearing around anions with weaker interactions decreases, causing the disappearance of the cages. For simplicity, the Ewald energy is used to evaluate the strength of electrostatic interactions between lithium ions and isolated anions. In Li8SiSe6_F(bar{4})3m, the two sites of Seiso have similar Ewald energy (−18.06 eV and −14.74 eV). However, in Li7SiSe5Cl, the results reveal that Cliso (−4.55 eV) has much higher Ewald energy than Seiso (−17.41 eV) and Li+ ions tend to distribute around Seiso rather than Cliso. Additionally, here is another typical example, two different isolated anions, Iiso and Siso, exist in solid electrolyte Li7P2S8I42,43, in which the Ewald energy of Iiso (−2.61 eV) is much higher than Siso (−15.96 eV), leading to the formation of Li+ cage pathways only surround Siso according to the Li+ trajectories revealed by AIMD at 600 K (Supplementary Fig. S9). The Ewald energy of isolated anions can be used as a practical parameter to determine in which part the cage pathways can be formed if multiple types of isolated anions exist in the structure. It can be served as the indicator to design which element should be doped to adjust the connectivity of the cages by modulating the interactions between Li+ ions and different types of isolated anions.
According to above characteristics summarized from Li8SiSe6, we deduce that the existence of isolated anions in structures is one of the ways to trigger cage transport mechanism of Li+ ions, the part of face sharing between neighboring cages can further reduce the barrier of inter-cage transport and result in high ionic conductivity. Six structures of Li8SiSe6 with various space group and Li7SiSe5Cl_F(bar{4}) 3m are taken as examples to analyse the factors influencing cage transport. First, the symmetry of isolated anions’ local structures affects intra-cage transport of Li+ ions, and the high symmetry enables the presence of more similar energy sites of Li+ around isolated anions and thus produce the frustration phenomenon. Second, the arrangement of isolated anions plays an important role in the inter-cage transport of Li+ ions. The densely packed isolated anions are more likely to produce cages with sharing faces, thus eliminate the limiting step of the inter-cage transport. Finally, when multiple types of isolated anions exist, the competition of the interaction between Li+ and different isolated anions affects the connectivity of the cages. The Ewald energy can help us to judge around which anions the cage pathways may be connected. We also attempted to establish quantitative relationships between the above variables and ionic conductivity (as shown in Supplementary Fig. S10 and Supplementary Note 4), but due to limited data points, it is still not sufficient to obtain reliable and general conclusions. The transport mechanism related to the isolated anions can be applied to find new fast ion conductors. By conducting the high-throughput screening from crystal structure databases based on the features of isolated anions, we have identified several types of fast ion conductors and the studies on their ion migration properties are carried out.
New conductors obtained by high-throughput screening
Since argyrodites with Li cages around isolated anions behave well in ionic transport, the high-throughput screening44 is performed to look for the crystal structure frameworks those share the isolated anion feature45. The structures in the Inorganic Crystal Structure Database (ICSD)46 and Materials Project (MP)47,48 database are considered. Four main criteria of the screening process are illustrated in Fig. 4a. Only the Li-containing compounds are considered. As a verification of the findings on isolated anions, we limit the screening process in ternary and quaternary compounds. Structures composed of more elements can be investigated using the same methodology. The Ehull values of the candidates are less than 15 meV/atom to keep the possibility for synthesis. Among them, the structures with isolated anions are recognized by bonding analysis and picked up for kinetic property simulations. Through these criteria, four types of structures with different conducting related isolated anions are obtained, including six compounds: Li6NI3, Li7N2I, Li5CrCl8, Li6VCl8, Li5SbS3I2 and Li8TiS6, as presented in Fig. 4. Each type of materials contains one of the isolated anions related to ion transport: N3-, Cl–, I– or S2-, and has its own structural characteristics. The point group of isolated anions’ local structure and the adjacent distances between them have been extracted for these structures. The former is identified to evaluate the intra-cage transport performance of the structure, and the latter generally relates to the inter-cage transport and acts as the limiting factor for ion transport. The information of isolated anions in conductors, including their element type, their local structures’ point group as well as distances between neighboring isolated anions is illustrated in Table 2. And the conductors’ structural and ion conducting properties obtained by AIMD are shown in Fig. 4 and Supplementary Fig. S11.
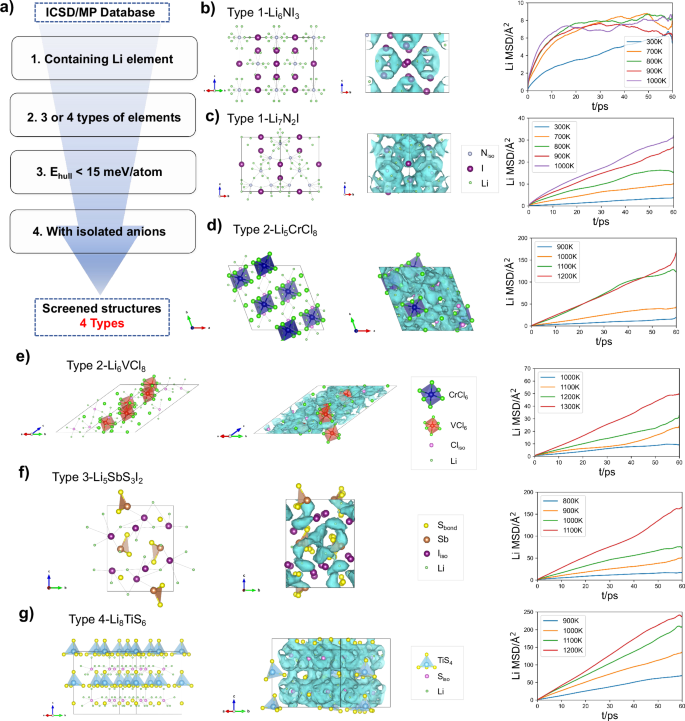
a High-throughput screening process looking for Li+ ion conductors with structural features of isolated anions. b–g Structures, Li+ diffusion pathways and MSDs of new conductors calculated by AIMD for Li6NI3, Li7N2I, Li5CrCl8, Li6VCl8, Li5SbS3I2 and Li8TiS6 respectively.
The first type relates to the N3− anions. Li6NI3 and Li7N2I, as shown in Fig. 4b, c, have similar structural frameworks possessing an alternating distribution of isolated I− and N3− atoms. We notice that Li7N2I with 0.5% carbon nanotube compounds was recently discovered experimentally and has a high ionic conductivity of 3.1 × 10−4 S/cm49, and many studies have also reported Li-N-X (X = I, Cl or S) electrolytes with isolated N3− ions50,51,52, ensuring the effectiveness of the screening. In these structures, Li+ ions are primarily distributed around N3− for the reason that the Ewald energy of Li+−N3− interaction (−33.27 eV in Li6NI3 and −48.88 eV in Li7N2I) is lower than that of Li+−I− interaction (−5.25 eV in Li6NI3 and −4.22 eV in Li7N2I). In Li7N2I, the point group symmetry of the isolated N3− ions’ local structure is Cs, which is lower than that of Li6NI3, Oh, but the distance between adjacent isolated N3− ions (3.85 Å) is quite smaller than that in Li6NI3 (6.32 Å). It can be inferred that compared with Li6NI3, Li7N2I has a worse intra-cage transport of Li+ ions, but a better inter-cage transport. The AIMD results further confirm the above inference. First of all, in both structures, the cage transport exists around N3- ions even at the AIMD simulations at 300 K, indicating that the frustration phenomenon is successfully formed because of the isolated anions. Only intra-cage transport of Li+ ions is found in Li6NI3 according to the fact that its MSDs at different temperatures (300 K, 700 K, 800 K, 900 K and 1000 K) have almost the same upper limit, and there is no connection between neighboring cages observed according to the trajectories of Li+ ions, which means Li6NI3 has not achieved long-distance diffusion, and calculating its diffusivity and ionic conductivity is meaningless, so the diffusivity and ionic conductivity for Li6NI3 in Table 2 are not given. However, for Li7N2I, it has lower MSD at 300 K (mainly low-barrier intra-cage transport) but much larger MSDs at higher temperatures (achieving inter-cage transport). The worse intra-cage transport in Li7N2I is due to the lower symmetry of isolated N3−’s local structure and the easier inter-cage transport is attributed to the reduction of distances of adjacent isolated N3− ions compared with Li6NI3. The face sharing Li cages in Li7N2I is also manifested by Li+ diffusion pathway. In addition to Li6NI3 and Li7N2I, the other screened structures containing isolated N3− anions are listed in Supplementary Table S1.
The second type is Li5CrCl8 and Li6VCl8 (Fig. 4d, e) containing isolated Cl– anions. In Li6VCl8, isolated Cl– anions’ local structure point group symmetry is Td, higher than that of C2v in Li5CrCl8, but the isolated Cl– anions are more loosely packed than in Li5CrCl8, indicating better intra-cage transport and worse inter-cage transport compared with Li5CrCl8, which is similar to the case in Li6NI3 and Li7N2I. But unfortunately, the obvious Li+ migration in these structures can only occur in AIMD at temperatures above 900 K. Although higher ionic conductivity is predicted in Li5CrCl8 than in Li6VCl8, the high barriers of the inter-cage transport act as the control factor of ion transport in both compounds. Furthermore, the other screened compound Li6NiCl8 is also listed in Supplementary Table S1.
The third type is Li5SbS3I2 (Fig. 4f) with isolated I– anions and it shows a relatively high conductivity. In this structure, the symmetry of I− ions’ local environment is Cs and the distance between Iiso is 4.02 Å and 4.36 Å. Although the structural characteristics meet the criteria, the complete cage-like pathways are not observed in the trajectories of AIMD. Upon closer examination of its crystal structure, it can be observed that the presence of [SbS3]3− groups disrupts the formation of cage channels. There is no tetrahedron formed by Sb3+ and S2− and the distance between Sb3+ and I− is only 3.70 Å, so the existence of Sb3+ around isolated I− sets up obstacles for creating complete cage-like pathways. But the low-barrier inter-cage transport, arising from the close proximity of isolated I− anions, provides a favorable pathway for the Li+ ion migration. Therefore, the element types of the surrounding ions of isolated anions can also affect the appearance of the cages. It also tells us additional information that the appearance of the pathways around the isolated anions can be adjusted through changing the position and environment of non-Li cations.
The fourth type is Li8TiS6 (Fig. 4g), it has the same structure as Li8SiSe6_hcp. It does not exhibit Li+ migration in AIMD simulations at 300 K, and the ion conductivity at 300 K is obtained through extrapolation of ionic conductivities at high temperatures.
The successful detection of the four types of Li conductors with isolated anions of N3−, Cl−, I−, and S2− confirms the feasibility of utilizing isolated anions as a screening feature. In addition, the anionic lattices of the above new conductors are not bcc but fcc, hcp or simple cubic (sc) structures as Table 2 shows, it implies that isolated anions can be introduced to structures with non-bcc anionic lattices to improve ion migration. And the influencing factors derived from argyrodite Li8SiSe6 are universally applicable to the analysis of pathways in such structures.
Discussion
In this study, we propose that isolated anions can serve as a structural feature for the formation of cage-like transport pathways, through which frustration phenomenon always appears and high ionic conductivity will be expected. Therefore, introducing and appropriately increasing the proportion of isolated anions in structures can be one of the methods to improve the ionic conductivity. Compared with bonded anions, isolated anions always show PDOS overlapping only with electronic states of Li+ ions and higher vacancy formation energy, which reveals their unique bonding and high stability.
By researching six Li8SiSe6 structures with different space group and Li7SiSe5Cl with F(bar{4}) 3m group, three factors related to isolated anions those influence Li+ ion transport are derived. To begin with, the symmetry of isolated anions’ local structure decides intra-cage transport and higher symmetry brings better intra-cage transport. Therefore, frustration phenomena can be created by introducing isolated anions and constructing their local environments with high symmetry. Following this principle, promising argyrodite ionic conductors can be predicted relying solely on structural regulation and this breaks the traditional idea of increasing argyrodites’ conductivity by stabilizing the cubic phase at room temperature through doping halogen elements. Besides, as for inter-cage transport, the arrangement of isolated anions becomes the most important factor because it is directly related to whether the cages are face-sharing connected or not, and the densely packed anions produce better inter-cage transport. This can explain the reason why some structures contain cage transport channels but can’t reach high conductivity. It also means that promoting the generation of face sharing cages through structural modification should be a promising strategy to improve ionic conductivity. Furthermore, when a structure owns several types of isolated anions, the interaction between different types of isolated anions and Li+ ions varies, thus affecting the connectivity inside the cage and the Ewald energy can help us quantify the impact and provide indications for element doping. All in all, the characteristics of isolated anions play a significant role in the discovery and design of ion conductors with frustration phenomena.
Additionally, by high-throughput method, four types of new Li+ ion conductors which contain isolated anions are identified. The ion transport behaviors in these conductors are demonstrated and analyzed by AIMD calculations, indicating that the principles derived from argyrodite Li8SiSe6 and Li7SiSe5Cl are suited generally for structures with the feature of isolated anions. Our conclusions and findings of isolated anions can be utilized for explaining a wide range of phenomena in structures with cage transport mechanism and can guide the design and improvement of these structures to reach higher ion conductivity. However, in what kind of structures isolated anions can be introduced and how the stability can be maintained are still open questions and further exploration is required.
Methods
Materials
Li6PS5Cl structure was obtained in Materials Project database (mp-985592). Six different structures of argyrodite Li8SiSe6 and the structure of Li7SiSe5Cl were obtained by atom substitution of other argyrodites in Materials Project database, ICSD database and the cif file of Li7Zn0.5SiS653 (Table 1). Li7P2S8I structure was obtained in ref. 43,44. For high-throughput search, the new conductors were screened from both Materials Project database and ICSD database. The structures were visualized by the VESTA software package54.
Density functional theory computation of structural relaxation and static calculations
We carried out all the relaxation calculations in primitive cells (Supplementary Table S2 lists the total number of atoms for each structure) using Vienna Ab initio Simulation Package (VASP)55 based on density functional theory (DFT) using Perdew–Burke–Ernzerhof (PBE)56 generalized gradient approximation (GGA) described by the projector-augmented-wave (PAW) approach. The cutoffs for the wave function and density are 520 eV and 780 eV, respectively. Both ions and cells were relaxed in the optimization, with the energy and force convergence criterion of 10−5 eV and 0.01 eV/Å, respectively. Spin-polarized DFT was used to relax Li5CrCl8 and Li6VCl8. The Ehull used to assess the thermodynamic stability was calculated using phase diagrams in the Materials Project (MP) database57,58. The point group of isolated anions’ local structure was determined by pymatgen.symmetry.analyzer.PointGroupAnalyzer Python package59. The Ewald energy used to determine the interactions between anions and Li+ ion was calculated by using the pymatgen.analysis.ewald Python packages.
Ab initio molecular dynamics simulations
We performed AIMD simulations to study ionic diffusion in supercell models (Supplementary Table S2 lists the total number of atoms for each structure) with lattice parameters around 10 Å, using nonspin-polarized DFT calculations with a Γ-centered k-point. Spin-polarized DFT was used to simulate Li5CrCl8 and Li6VCl8. The AIMD run was carried out with a Nose thermostat60 for 70,000 steps, with a time step of 1 fs. We picked different temperatures to simulate different materials. The first 10 ps is used for structural equilibrium and the last 60 ps is used for kinetic property analysis. The results of them were used to investigate the diffusivity, ionic conductivity as well as diffusion pathways. The ionic conductivity was calculated following the established method61. We used positions of high probability density of Li ions calculated as the fraction of time that each spatial location is occupied to describe the diffusion pathways of Li62. We performed the diffusion analysis by using pymatgen and the pymatgen.analysis.diffusion.analyzer Python packages63.


Responses