Potent and broadly neutralizing antibodies against sarbecoviruses elicited by single ancestral SARS-CoV-2 infection
Introduction
The relentless spread of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has led to millions of infections and substantial morbidity and mortality across continents, severely challenging healthcare systems, economies, and societal norms. The clinical spectrum of SARS-CoV-2 infection ranges from asymptomatic or mild respiratory illness to severe pneumonia, acute respiratory distress syndrome, multi-organ failure, and death, with certain populations, such as the elderly and those with underlying health conditions, being at higher risk of severe outcomes1. The genome of SARS-CoV-2 encodes several structural and non-structural proteins crucial for viral replication, transcription, and immune evasion. Among these proteins, the Spike (S) protein plays a pivotal role in viral entry by binding to the angiotensin-converting enzyme 2 (ACE2) receptor on host cells, facilitating membrane fusion, and initiating infection2,3. The S protein is a major target for host immune responses and serves as the primary antigen for vaccine development and immunotherapeutic interventions.
Monoclonal antibody (mAb) therapeutics targeting the S protein of SARS-CoV-2 represent a vital option for both prophylaxis and treatment, especially for individuals who are immunocompromised, unable to receive a vaccination or have other risk factors predisposing them to severe disease4. Throughout the course of infection, antibodies exert their therapeutic effects by various mechanisms, including neutralizing virions through blocking receptor binding, crosslinking viral proteins, inhibiting fusion with host cells, and facilitating immune clearance of virions and infected cells5 .
However, as SARS-CoV-2 continues to evolve, numerous variants carrying multiple mutations have emerged, resulting in significant evasion of antibody neutralization6,7,8,9. All anti-SARS-CoV-2 mAbs and mAb cocktail therapeutics previously approved for Emergency Use Authorization (EUA) by the FDA were subsequently withdrawn due to immune escape10, except for one exception—PEMGARDA (pemigard), which recently obtained EUA for pre-exposure prophylaxis (PrEP)11. Some early-discovered antibodies and their combinations, such as Bamlanivimab/Etesevimab, Casirivimab/imdevimab (REGN-COV2), and Tixagevimab/Cilgavimab (Evusheld), lost their neutralizing activity against SARS-CoV-2 variants9,12,13,14,15,16. Bebtelovimab (LY-CoV1404), while highly efficient in neutralizing many SARS-CoV-2 variants, including early Omicron subvariants, escaped neutralization by subsequently emerging BQ.1.1 and XBB lineages15,17. Another mAb isolated from a SARS-CoV convalescent individual, Sotrovimab (S309), maintained its neutralizing activity against most emerged SARS-CoV-2 variants due to recognition of a conserved epitope4,8,18,19. However, its neutralizing potency is mediocre, with an IC50 value of around 1 μg/ml against many SARS-CoV-2 variants15,19, leading to its EUA withdrawal as well. These findings underscore the imperative for the development of next-generation antibodies capable of effectively resisting immune evasion induced by SARS-CoV-2 evolution.
To date, some neutralizing antibodies with cross-neutralization activities against SARS-CoV-2 and its variants have been identified. One example is SA55, isolated from SARS-CoV-2-vaccinated SARS convalescents has been reported to effectively resist mutations that have emerged in SARS-CoV-220. Additionally, antibodies targeting the SD1 region, such as S3H3 and sd1.040, have demonstrated efficient cross-neutralization against major SARS-CoV-2 variants due to sequence conservation21,22,23. While the S2 subunit exhibits greater conservation compared to S1, antibodies targeting S2 typically exhibit suboptimal potency for achieving protection4,24,25,26. Recently, we reported the isolation of potent and broadly neutralizing antibodies from a vaccinated donor who received a special five-dose COVID-19 vaccination schedule. Among these, two mAbs, PW5-5 and PW5-535, were found to possess pan-sarbecovirus potential27. Most of the above-mentioned broadly neutralizing antibodies have been isolated from individuals who received multiple vaccinations, experienced multiple infections, or a combination of both. However, a few broadly neutralizing antibodies have also been reported from early infections with only the prototype SARS-CoV-2 virus28,29. In this study, we present what we believe to be the most potent and broadly neutralizing antibodies isolated from prototype infection, capable of neutralizing all currently known variants, including KP.2, KP.3, and XEC, to which the donor had not been previously exposed.
Results
SARS-CoV-2 and SARS-CoV cross-reactive antibodies from ancestral SARS-CoV-2 infection
We recruited three COVID-19 convalescents who had been infected with the original SARS-CoV-2 virus during the initial outbreak in China, a period when vaccines were not yet available (Fig. 1a). All participants had confirmed viral exposure, as evidenced by positive RNA tests, and had received treatment at Guangzhou Eighth People’s Hospital. In May 2020, four months after their infection, we collected peripheral blood mononuclear cells (PBMCs) from these individuals. Memory B cells (MBCs) were subsequently sorted, and all MBCs were cultured without bias towards any specific antigen (Supplementary Fig. 1). The responses of SARS-CoV-2 and SARS-CoV cross-reactive MBCs have been investigated, and the findings are forthcoming in another publication. From a single well of these cultured MBCs from Pt2, which demonstrated cross-reactivity to both SARS-CoV-2 and SARS-CoV, we cloned one antibody heavy chain and two light chains (Fig. 1b), which were then assembled into two human IgG1 antibodies, named CYFN1006-1 and CYFN1006-2.

a The clinical profiles of the three SARS-CoV-2 convalescent donors. b The gene usage and CDR sequences of the cloned mAbs exhibiting cross-activity against both SARS-CoV-2 and SARS-CoV. c Binding of the indicated mAbs to S, S1, S2, RBD, NTD of SARS-CoV-2 and S, RBD of SARS-CoV, as measured by ELISA. Data represented means ± SEM. d Binding affinity of the indicated mAbs to SARS-CoV-2 WT RBD, XBB.1.16 RBD, and SARS-CoV RBD was analyzed by BLI. N.B. no binding, N.A. not applicable, binding too weak to accurately calculate.
We assessed the binding activity of these two mAbs to SARS-CoV-2 and SARS-CoV, in comparison with SA55, a broad sarbecovirus neutralizing antibody isolated from SARS-CoV-2-vaccinated SARS convalescents20. An irrelevant antibody (3G2, a ZIKV NS1-specific Ab30) was included as a negative control. Both CYFN1006-1 and CYFN1006-2 demonstrated strong binding to the S1 subunit of the SARS-CoV-2 S trimer while showing no binding to the S2 subunit, as detected by ELISA assay. Within the S1 subunit, both mAbs bound to the RBD, but not to the NTD. Although CYFN1006-1 and CYFN1006-2 exhibited similar binding to SARS-CoV-2, their binding patterns to SARS-CoV S and RBD appeared different. Specifically, CYFN1006-1 showed robust binding to SARS-CoV S and RBD, whereas CYFN1006-2 displayed significantly weaker binding to the SARS-CoV antigens (Fig. 1c).
Their binding affinity to the RBDs of SARS-CoV-2 WT, XBB.1.16, and SARS-CoV was further quantified using Bio-Layer Interferometry (BLI), as illustrated in Fig. 1d. CYFN1006-1 exhibited very strong binding (<0.001 nM) to SARS-CoV-2 WT RBD, reduced but still substantial (1.37 nM) binding to SARS-CoV-2 XBB.1.6 RBD and further reduced (32.4 nM) binding to SARS-CoV RBD. Meanwhile, CYFN1006-2 also demonstrated very strong binding to SARS-CoV-2 WT RBD and reduced binding to XBB.1.6 RBD. However, its binding affinity to SARS-CoV RBD was diminished to a level too low to accurately measure. In contrast, SA55 exhibited the highest binding affinity to SARS-CoV RBD but reduced affinity to SARS-CoV-2 RBDs.
Potent and broad neutralizing activity of CYFN1006-1 and CYFN1006-2
Next, we sought to evaluate the breadth of neutralization achieved by these two antibodies. We compiled a comprehensive panel of 45 pseudoviruses, encompassing all representative SARS-CoV-2 variants, SARS-CoV, bat or pangolin coronaviruses in the SARS-CoV-2-related lineage (RaTG13, GD-Pangolin) and SARS-CoV-related lineage (WIV1, SHC014), as well as MERS-CoV (Fig. 2a; Supplementary Fig. 2). The SARS-CoV-2 variants in our panel included previously identified Variants of Concern (VOCs) (B.1.1.7, B.1.351, P.1, B.1.617.2, BA.1) and Variants of Interest (VOIs) (B.1.525, B.1.621, C.37, etc), comprising variants that were once prevalent such as BA.2, BA.2.12.1, BA.2.75, BA.4/5, BQ.1.1, CH.1.1, and various XBB subvariants, as well as those recently identified and currently circulating variants such as JN.1, KP.2, KP.3, KP.3.1.1 and XEC. The mutations dispersed across the virus S protein are depicted in Fig. 2b.

a Phylogenetic tree of the tested comprehensive panel of pseudotyped viruses, including SARS-CoV-2 variants, SARS-CoV, and their related sarbecoviruses, as well as MERS-CoV. b Matrix of mutations on S protein across SARS-CoV-2 variants. c Neutralization IC50 values of the six indicated mAbs against the comprehensive panel of pseudotyped viruses.
We assessed the neutralization potency of CYFN1006-1 and CYFN1006-2 on this panel of pseudoviruses, with four previously reported mAbs (SA55, S309, S3H3, and LY-CoV1404) for comparison. As previously reported, LY-CoV1404 completely lost its activity against descendants of BQ, CH, and XBB, as well as the JN.1, KP.2, KP.3, and XEC variants. The SD1-targeting mAb S3H3 was rendered completely inactive against the BA.2.86 and JN.1 variants, likely due to the presence of the E554K mutation in the antibody’s epitope. S309 retained activity, albeit significantly reduced, against most post-Omicron variants, but it completely lost activity against BN.1, KP.2, KP.3, and XEC variants, likely due to the effects of the K356T mutation (Fig. 2b, c).
In contrast, CYFN1006-1 demonstrated consistent neutralization of all tested SARS-CoV-2 variants with unaffected potency, displaying IC50 values ranging from approximately 1 to 5 ng/mL, comparable to or even superior to those of SA55. Similarly, CYFN1006-2 exhibited high potency against all SARS-CoV-2 variants, with a slightly reduced efficacy against KP.2. In terms of their activity against the extended sarbecoviruses, both CYFN1006-1 and CYFN1006-2 efficiently neutralized SARS-CoV-2-related viruses (RaTG13, GD-Pangolin). However, only CYFN1006-1 maintained reduced neutralization potency against SARS-CoV and the SARS-CoV-related lineage (WIV1, SHC014), while CYFN1006-2 did not neutralize SARS-CoV or WIV1. As anticipated, none of these monoclonal antibodies neutralized MERS-CoV (Fig. 2c).
Given the outstanding neutralization activity of CYFN1006-1 and CYFN1006-2 against pseudoviruses, we extended our testing to authentic viruses, including various SARS-CoV-2 variants (WT, BA.1, XBB.1, EG.5.1, BA.2.86, and JN.1) and SARS-CoV (GZ50). Consistent with the pseudovirus results, both mAbs effectively neutralized all tested SARS-CoV-2 variants with similarly high potency. For SARS-CoV, CYFN1006-1 demonstrated the ability to completely neutralize the virus, albeit with less potency, whereas CYFN1006-2 did not exhibit neutralizing activity against it (Supplementary Fig. 3). Taken together, these results confirm that a single ancestral SARS-CoV-2 infection could elicit antibody response against conserved epitopes, albeit rare, with broad-spectrum neutralization activity against evolving viral variants that the donor had never been exposed to.
Cryo-EM structure of CYFN1006-1 complexed with SARS-CoV-2 XBB.1.16 S trimer
We, therefore, next explored the structural basis of the broad neutralization activity of CYFN1006-1 and CYFN1006-2 and tried to explain the mechanism of their difference in neutralization. First, the structure of CYFN1006-1 immunoglobulin G (IgG) in complex with the SARS-CoV-2 XBB.1.16 S was determined using cryo-electron microscopy (cryo-EM). The XBB.1.16 S trimer was mixed with CYFN1006-1 at a molar ratio of 1:1.2, incubated at 4 °C for 30 mins, and purified by gel filtration chromatography. The XBB.1.16 S-CYFN1006-1 complex was used for the cryo-EM study (Supplementary Fig. 4).
Cryo-EM analysis has unveiled three distinct conformational states of the XBB.1.16 S-CYFN1006-1 complex (Fig. 3a; Supplementary Fig. 5). In state 1 (~2% of the particles), two S trimers with ‘3 down-RBDs’ each interact with three CYFN1006-1 molecules, forming a di-trimer complex (5.41 Å). In state 2 (~36% of the particles), XBB.1.16 S trimers with ‘3 down-RBDs’ bind three CYFN1006-1s (2.87 Å). Approximately, 62% of the particles adopt the state 3 conformation, where XBB.1.16 S trimers with ‘2 down-RBDs’ bind with three CYFN1006-1s (2.84 Å). The S RBD with CYFN1006-1 Fab region was locally refined to 2.92 Å. The Fc region is not visible in the final structure due to its flexibility.

a Cryo-EM structure of the XBB.1.16 S trimer in complex with CYFN1006-1 IgG, shown in surface. b Structure of XBB.1.16 RBD-CYFN1006-1. The RBD is displayed in orange surface mode. The heavy chain and light chain of CYFN1006-1 are shown as ribbons colored in sea green and green yellow, respectively. ACE2 binding epitopes are shown in yellow. c Superimposition of RBD-CYFN1006-1 with RBD-ACE2 (PDB: 7XAZ) reveals the relative position of ACE2 and CYFN1006-1. d Top view of S trimer complexed with CYFN1006-1s, showing that RBDs interact with the neighboring CYFN1006-1. The RBD and CYFN1006-1 residues involved in the interactions are colored green and red, respectively. e Detailed interactions between the CYFN1006-1 and XBB.1.16 RBD. Hydrogen bonds are represented by dashed lines. Hydrophobic interactions are marked with red circles.
CYFN1006-1 binds to the outer side of RBD, burying a surface area of 933 Å2. The epitopes are positioned next to the receptor-binding motif (RBM) of ACE2 (Fig. 3b). Structural superposition of the RBD-ACE2 (PDB: 7XAZ) and XBB.1.16 RBD-CYFN1006-1 complex indicates that CYFN1006-1 is close to the glycan on ACE2N53 but does not produce steric hindrance conflict with ACE2 (Fig. 3c). This is consistent with the ACE2 binding test, which showed that CYFN1006-1 did not prevent the binding between ACE2 and RBD (Supplementary Fig. 6). The CYFN1006-1 Fabs on the S trimer can interact with the adjacent Fabs through hydrogen bonding and van der Waals forces between heavy chains (Supplementary Fig. 7a). In addition, D62 and S63 of CYFN1006-1 heavy chain can form hydrogen bonds with T500 of the adjacent RBD (Supplementary Fig. 7b), locking the RBD in the “down” conformation. Five of six complementary determining regions of CYFN1006-1 are engaged in RBD binding, involving a total of 17 RBD residues (Fig. 3d; Supplementary Fig. 8). A rich interaction network was formed, including 18 pairs of hydrogen bonds and 1 hydrophobic interaction patch. V30 of CDRL1 can interact with E340, V341, F347, N354 and K356 of RBD. Y107, A108, S114 of CDRL3 and D112A, and W112 of CDRH3 can form 7 pairs of hydrogen bonds with RBD residues N343, A344, and T345. The D62 and D64 of CDRH2 form two pairs of hydrogen bonds with the K440 of RBD. D109-W112 of CDRH3 can form 5 pairs of hydrogen bonds with S446, K444, and T346. Notably, N343, A344, and T345 play an important role in stabilizing CYFN1006-1 binding to RBD and are highly conserved in various mutant strains, which are important sites for antibody drug development and vaccine design. K440, L441, P445, and P499 of RBD are embedded in the hydrophobic patch formed by CDRH1 (Y36, Y37, W38) and CDRH3 (W112) to further stabilize the CYFN1006-1 binding to RBD (Fig. 3e). In addition, S399 and R509 of RBD are also involved in CYFN1006-1 binding through van der Waals forces.
Cryo-EM structure of CYFN1006-2 complexed with SARS-CoV-2 EG.5.1 S trimer
In addition, we used cryo-EM to analyze the structure of the EG.5.1 S-CYFN1006-2 complex using the same method as the XBB.1.16 S-CYFN1006-1 complex. Cryo-EM analysis revealed two conformations of the EG.5.1 S-CYFN1006-2 complex. Approximately, 2% of the particles consist of EG.5.1 S trimers with ‘2 down-RBDs’ bound to three CYFN1006-2 molecules (3.63 Å). Most particles (~92%) form di-trimers, where two S trimers with “3-down RBD” bind three CYFN1006-2 molecules to assemble a di-trimer (Fig. 4a), resembling the CYFN1006-1 state1 confirmation. One S trimer with three CYFN1006-2 Fab is locally refined to 3.04 Å, and the RBD-CYFN1006-2 Fab region was locally refined to 2.96 Å (Supplementary Fig. 9).
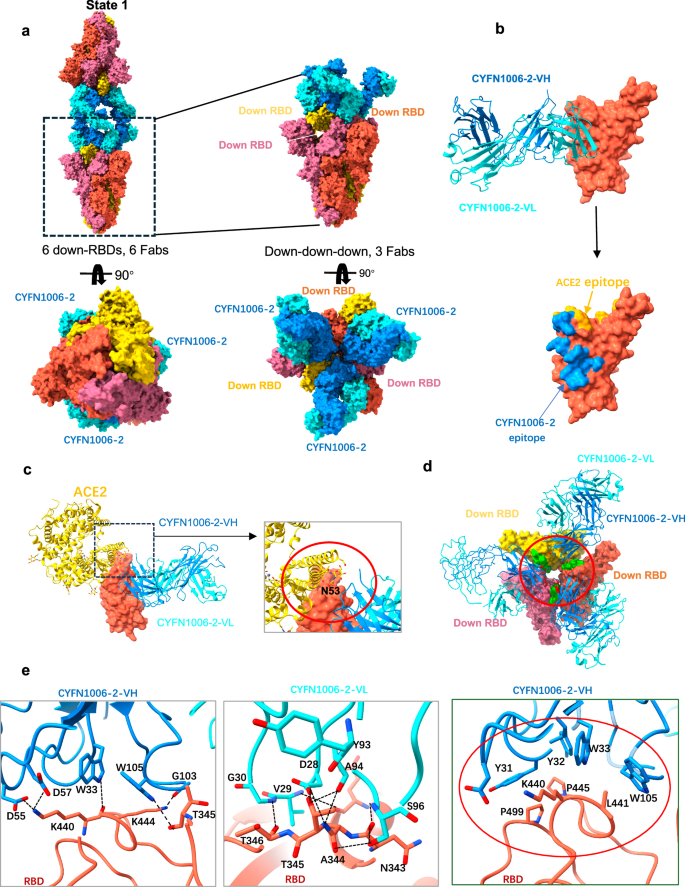
a Cryo-EM structures of the EG.5.1 S trimer in complex with the CYFN1006-2 IgG, shown on the surface. b Structure of EG.5.1 RBD-CYFN1006-2. The RBD is displayed in orange surface mode. The heavy chain and light chain of CYFN1006-2 are shown as ribbons colored in dodger blue and cyan, respectively. ACE2 binding epitopes are shown in yellow. c Superimposition of RBD-CYFN1006-2 with RBD-ACE2 (PDB: 7XAZ) revealed the relative position of ACE2 and CYFN1006-2. d Top view of S trimer complexed with CYFN1006-2s, showing that RBDs interact with the neighboring CYFN1006-2. The RBD and CYFN1006-2 residues involved in the interactions are colored green and red, respectively. e Detailed interactions between the CYFN1006-2 and EG.5.1 RBD. Hydrogen bonds are represented by dashed lines. Hydrophobic interactions are marked with red circles.
Structural analysis shows that the epitopes of CYFN1006-2 on S RBD are very similar to those of CYFN1006-1. When compared to CYFN1006-1, the epitopes of CYFN1006-2 are slightly shifted away from RBM and do not interact with R509 next to RBM. The binding surface area is 875 Å2, involving a total of 16 amino acids, forming 14 pairs of hydrogen bonds and a hydrophobic plaque (Fig. 4b). Structural superposition of the RBD-ACE2 (PDB: 7XAZ) and EG.5.1 RBD-CYFN1006-2 complex indicates that the CYFN1006-2 is adjacent to the glycosylated residue ACE2N53, but does not clash with ACE2 (Fig. 4c). This is also consistent with the ACE2 binding test (Supplementary Fig. 6). Three CYFN1006-2 Fabs bound to the same S trimer with the heavy chains interacting with each other by van der Waals forces. In addition, the binding mode of CYFN1006-2 is similar to that of CYFN1006-1, S63 of each heavy chain can form hydrogen bonds with T500 of the adjacent RBD, preventing the RBD from forming “up” conformation (Fig. 4d; Supplementary Fig. 7c). K440, L441, P445, and P499 of RBD are embedded in the hydrophobic bags of CDRH1 (Y36, Y37, W38) and CDRH3 (W112) to further stabilize the combination of CYFN1006-2 and RBD. N343, N344, and T345 of RBD are highly conserved in a variety of SARS-CoV-2 mutants and can form eight pairs of hydrogen bonds with D29 of CDRL1, Y107, A108, S114 of CDRL3, and W112 of CDRH3, which may be the reason for the broad-spectrum neutralization of CYFN1006-2. K440, K444, and T346 participate in the formation of five pairs of hydrogen bonds (Fig. 4e). In addition, RBD residues H339, E340, V341, F347, N354, S399, and S446 are also involved in CYFN1006-2 binding through van der Waals forces.
Therefore, CYFN1006-2 can neutralize SARS-CoV-2 by similar mechanisms as CYFN1006-1, such as maintaining the RBD in the down conformation and crosslinking S trimers to di-trimers. However, what makes the difference between CYFN1006-1 and CYFN1006-2 in neutralizing SARS-CoV? Since the binding affinity of CYFN1006-2 to SARS-CoV RBD is too low, we superimposed SARS-CoV RBD (PDB: 2DD8) with SARS-CoV-2 RBD to predict the interactions between SARS-CoV RBD and CYFN1006-1/CYFN1006-2. Structural analysis revealed that CYFN1006-1 may form three more pairs of hydrogen bonds with SARS-CoV RBD than CYFN1006-2 (Supplementary Fig. 10a). This finding is consistent with and can explain the binding and neutralization results, which demonstrated that CYFN1006-1 bound to and neutralized SARS-CoV much more effectively than CYFN1006-2. Furthermore, our results also showed that the main difference in the neutralizing activity of CYFN1006-1 and CYFN1006-2 among the SARS-CoV-2 variants was against the KP.2 variant, with CYFN1006-2 showing significantly weaker activity than that of CYFN1006-1. We predicted KP.2 RBD-antibody complex models, showing that CYFN1006-1 could form more hydrogen bonds with KP.2 RBD than CYFN1006-2 (Supplementary Fig. 10b). The R346T mutation, present in KP.2 but not in KP.3, causes the RBD to lose hydrogen bonds with E56 of CYFN1006-2-VL and 112 A of CYFN1006-2-VH (Supplementary Fig. 10c). This explains the weakened neutralization of CYFN1006-2 against KP.2, but not against the more recent KP.3 and KP.3.1.1 variants. The sequence difference between CYFN1006-1 and CYFN1006-2 is located in the light chain CDR1 (I26A, D36Y) and CDR2 (D56E, S65N). Although these sites do not directly participate in binding with the RBD, we found that I26, D36, and D56 of CYFN1006-1 form hydrogen bonds with their corresponding residues (N85, N37, and Y107) in the light chain, while these hydrogen bonds are lost in CYFN1006-2 (Supplementary Fig. 10d). The presence of these additional hydrogen bonds in CYFN1006-1 is speculated to contribute to its more stable structure, thereby enhancing its neutralization effect against SARS-CoV and KP.2.
Protection of CYFN1006-1 against SARS-CoV-2 Omicron JN.1 in hamsters
The in vitro neutralization results indicated that, compared to CYFN1006-2, CYFN1006-1 exhibited similar activity against all the tested SARS-CoV-2 variants but demonstrated a broader neutralization breadth to cover SARS-CoV and related sarbecoviruses. Therefore, we selected CYFN1006-1 to assess its anti-SARS-CoV-2 activity in vivo using a hamster model. Male hamsters (aged 6–8 weeks) were intranasally challenged with SARS-CoV-2 Omicron JN.1 at 8 h after being treated with one dose of CYFN1006-1 antibodies (20 mg/kg) through intraperitoneal injection. At day 4 post-virus inoculation, the hamsters were sacrificed to harvest nasal turbinate (NT) and lung tissues for virological assessments (Fig. 5a). Our results demonstrated that passive immunization with CYFN1006-1 significantly reduced the replication of JN.1 in the hamster lungs, as evidenced by the significantly lowered viral subgenomic E (sgE) gene copy and infectious virus titers (Fig. 5b). In parallel, we performed immunofluorescence staining and histopathological analysis of SARS-CoV-2 nucleocapsid (N) protein in the harvested hamster lungs. Compared with the PBS-treated group, the expression of viral N protein was substantially reduced in the lungs of infected hamsters upon CYFN1006-1 treatment (Fig. 5c). Importantly, histopathological examination of the lung showed multi-focal inflammation, collapse, and congestion of the alveoli wall, epithelial damage in the small bronchioles, and alveolar collapse in the PBS-treated hamsters. In contrast, these histological changes were markedly attenuated in the lungs of CYFN1006-1-treated hamsters (Fig. 5d). Due to the self-limiting nature of infection in the hamster model, no significant differences in body weight change were observed between the groups (Supplementary Fig. 11). Overall, our results reveal that CYFN1006-1 have the potential to neutralize SARS-CoV-2 Omicron JN.1 in vivo.

a Schematic of the in vivo mAb efficacy test experiments in hamsters. 6-to-8-week-old male Syrian golden hamsters were treated with one dose of mAb CYFN1006-1 antibody (20 mg/kg, n = 5) through intraperitoneal injection antibodies at 8 h before challenging with 1 × 104 PFU JN.1. Animals were euthanized at 4 dpi for collection of nasal turbinates (NT) and lung tissues for detection of viral burden and viral antigen expression. b, c Hamster NT and lungs viral sgE copies and infectious titers were measured with (b) probe-specific RT-qPCR and (c) TCID50 assay. d SARS-CoV-2-N protein in hamster lung tissue was stained with anti-SARS-CoV-2-N antibody and visualized using fluorescent microscopy, with DAPI staining used for cell nuclei. Scale bar, 200 μm. e Histological examination of hamster lungs with hematoxylin and eosin (H&E) staining. Scale bar, 200 μm. Data represented means ± SEM from the indicated number of biological repeats. p Values were determined by unpaired t-test.
Discussion
Neutralizing antibodies with robust autologous neutralization activity often exhibit reduced effectiveness against future variants, especially those obtained early in the pandemic from individuals infected or vaccinated with the ancestral strain. To date, the majority of antibodies exhibiting cross-neutralization activities have been isolated from donors who have experienced prior SARS infection, multiple vaccinations or infections, or possess hybrid immunity resulting from both vaccinations and infections20,27. In this study, we have successfully identified antibodies capable of potently neutralizing all major SARS-CoV-2 variants, originating from a human donor with ancestral virus exposure exclusively. Notably, considering the timing of sample collection and the donor’s age, it is highly unlikely that this individual had prior exposure to either the SARS-CoV-2 vaccine or SARS-CoV infection. From this donor, we cloned two antibodies, both bearing the rare IGHV5-51 heavy-chain gene but paired with different light-chain genes. Interestingly, while these antibodies demonstrated similarly high potency in neutralizing SARS-CoV-2 variants, they exhibited varying degrees of efficacy in neutralizing SARS-CoV and related animal sarbecoviruses.
In our study, we employed an unbiased antibody discovery method31, which involved culturing all MBCs without specific bait, followed by screening the culture supernatant for cross-activity against both SARS-CoV-2 and SARS-CoV. Through this approach, we successfully identified two antibodies with cross-neutralization activity from individuals with solely ancestral virus exposure. Notably, these antibodies were isolated from a single well, demonstrating potent and broad neutralization against both SARS-CoV-2 and SARS-CoV, out of more than 2000 wells, underscoring the rarity of such antibodies. Our findings highlight the feasibility of discovering rare yet broad-spectrum neutralizing antibodies capable of effectively targeting future circulating strains, even in convalescent patients who were infected with the prototype strain.
We’ve uncovered the structural foundation behind the broad neutralization activity of these antibodies. The binding epitopes of CYFN1006-1 largely overlap with LY-CoV140417, REGN1098712, and S30918, situated on the outer surface of RBD near the RBM region (Supplementary Fig. 12). The epitopes comprise the important residues of F340, N343, T345, R346, K356 in S309 epitope, forming numerous hydrogen bonds and displaying high conservation. The epitope recognized by CYFN1006-1 shares 10 amino acids with CV38-14232, which also utilizes the same IGHV5-51 gene as CYFN1006-1. However, the neutralization of CYFN1006-1 against SARS-CoV-2 (IC50 < 5 ng/mL) is much more potent than that of CV38-142 (IC50 = 3.46 μg/mL). This difference could be explained by the larger epitope region targeted by CYFN1006-1 on the RBD compared to that of CV38-142. In contrast to LY-CoV1404, CYFN1006-1 lacks high-frequency mutation sites such as N439 and N501, rendering its epitopes more conservative. CYFN1006-1 exerts its neutralizing effect not only by locking RBDs in the down position but also by aggregating virions through cross-linking S trimers to form di-trimers. When CYFN1006-1 binds to S of “3 down-RBDs “, the heavy chains of the adjacent Fab interact with each other and can form hydrogen bonds with the adjacent RBD, locking the RBD in the “down-RBD” conformation. Although other antibodies, such as 6M633, 553-1534, and Fu235, have also been reported to induce di-trimer formation, their binding epitopes and the mode of di-trimer formation differ significantly from those of our antibodies (Supplementary Fig. 13). The di-trimers formed by 6M6, 553-15, and FU2 exhibit “6 up-RBDs, 6 Fabs”, with two trimers cross-linked into di-trimers through RBD-RBD interactions and the binding of antibodies to neighboring RBDs. In contrast, the di-trimers formed by CYFN1006-1/2 consist of “6 down-RBDs, 6 Fabs”, cross-linked through antibody interactions or Fc interactions.
The binding epitopes and neutralization mechanism of CYFN1006-2 closely resemble those of CYFN1006-1, albeit with a slight downward shift. However, it is important to acknowledge that different S proteins were used in the structural analysis: CYFN1006-1 was analyzed with XBB.1.16 S, while EG.5.1 S was used for CYFN1006-2. This difference in spike protein usage represents a limitation of this study. Ideally, it would be preferable to compare the structures of each antibody bound to the same S protein. Nonetheless, the structural comparison suggests that broadly neutralizing antibodies could target shared hotspot epitopes while contacting residues resistant to mutations, thereby maintaining their efficacy against evolving viral strains.
SA55 was isolated from SARS survivors vaccinated with the SARS-CoV-2 vaccine and exhibited higher neutralization activity against SARS-CoV compared to our antibodies, particularly CYFN1006-2. Structural analysis revealed that SA55’s binding epitopes are more conserved between SARS-CoV and SARS-CoV-2, and it forms more hydrogen bonds with the SARS-CoV RBD than CYFN1006-2 (Supplementary Fig. 14). Notably, both SA55 and CYFN1006-1 may cover all clinical related viral strains evolved so far (Fig. 2). CYFN1006-1 shares two amino acids (P445, P499) with the SA55 epitope20, leading to spatial conflict and preventing simultaneous binding to the RBD, as confirmed by competition ELISA tests (Supplementary Fig. 15). However, SA55 and CYFN1006-1 bind largely different conserve region of RBD (Supplementary Fig. 12). SA55 directly competes RBD binding to ACE220, while CYFN1006-1 does not (Supplementary Fig. 6). Therefore, a cocktail with combination of SA55 and CYFN1006-1 may still benefit in covering future evolving mutants of SARS-CoV-2 than individual antibodies if used as drugs. A cocktail of three distinct potent and broad neutralizing mAbs may be required to maintain efficacy against evolving viral strains in the future and to reduce the potential risk of the emergence of resistant variants associated with the use of single mAb therapies36,37.
Furthermore, in vivo studies in a hamster model demonstrated the protective efficacy of CYFN1006-1 against SARS-CoV-2 infection, providing additional evidence of its therapeutic potential. Overall, these findings emphasize the critical need for continued investment in antibody discovery and development programs to address the ongoing challenges posed by the dynamic nature of the SARS-CoV-2 virus and other related coronaviruses.
Method
Peripheral blood mononuclear cell (PBMC) samples
This study was performed with the approval of the Institutional Review Boards (IRB) at the Guangzhou Eighth People’s Hospital (20200134). All ethical regulations relevant to human research participants were followed. Blood samples were drawn after each participant provided a written informed consent form. Fresh PBMCs from three COVID-19 patients after four months of SARS-CoV-2 infection were used for B cell isolation using fluorescence-activated cell sorting (FACS). All patients had positive RNA results for SARS-CoV-2 infection and were hospitalized at Guangzhou Eighth People’s Hospital, Guangzhou Medical University, China, in January and February 2020, the early outbreak of COVID-19 in China.
Expression of S proteins and their subdomain proteins
The proteins for epitope mapping of the isolated antibody were expressed in mammalian cells. Briefly, the coding sequence of the S ectodomain of SARS-CoV-2 (NC_045512.2) or SARS-CoV (NC_004718.3) was inserted into pcDNA3.1 expression vector (Invitrogen, Waltham, MA) with a D7-tag on the C-terminus of each S protein gene as previously reported31. Similarly, the expression vectors for S subdomain proteins (S1, S2, RBD, NTD for SARS-CoV-2 and RBD for SARS-CoV) were also constructed. The recombinant plasmid was transfected into mammalian HEK293F cells. After culturing, S protein-containing supernatants were collected and clarified by centrifugation. Aliquots of supernatants were added with a protease inhibitor and then stored at −80 °C until use.
Isolation and cloning of mAbs
Patient-derived mAbs against SARS-CoV-2 S protein were generated from memory B cells (CD19+IgD−IgM−CD27+CD38low) in the PBMCs from COVID-19 patients using an in vitro B cell stimulation approach as previously reported38. Briefly, memory B cells in fresh PBMCs from convalescent COVID-19 patients were sorted into 96-well culture plates with 50 cells per well. B cells were cultured for 7–10 days in a culture medium supplemented with growth factors and stimuli including IL-21, Chk2 inhibitor, and CpG2006 in the presence of feeder cells (irradiated PBMCs from healthy blood donors). The culture supernatants were screened for S protein binding using a capture ELISA. The ELISA microplates precoated with anti-D7 tag Ab (Excellgen, EG-283, 4 μg/ml) were used to capture the S protein with D7-tag in the supernatant of HEK293F cells transfected with recombinant pcDNA3.1. Cells in the positive wells were subjected to RNA extraction for cDNA synthesis using the SuperScript™ III First-Strand Synthesis System (Invitrogen, Waltham, MA). The cDNA was used as a template of the nested PCR with gene-specific primers or primer mixes to amplify the transcripts of antibody heavy and light variable genes as previously reported39. The PCR products were purified from agarose gel using a QIAEX II Gel Extraction Kit (Qiagen, Hilden, Germany). Purified PCR products were digested with restriction enzymes AgeI/SalI, AgeI/BsiWI, or AgeI/XhoI (ThermoFisher Scientific, Waltham, MA) and followed by ligation into human IgG1, Igκ, or Igλ expression vectors, respectively. The recombinant Ab-expressing constructs were sequenced and analyzed using the IMGT/V-QUEST (IMGT, France), a sequence alignment software for the immunoglobulin sequences of the variable regions.
Epitope mapping
For epitope mapping of the isolated mAbs, a panel of SARS-CoV-2 and SARS-CoV S, and their subdomain proteins (S1, S2, RBD, NTD for SARS-CoV-2 and RBD for SARS-CoV) were used. The binding of each mAb to these proteins was measured using a capture ELISA. Human mAbs starting at 16 µg/ml were used for preparing a 4-fold serial dilution and applied to the capture ELISA. All samples were detected in duplicate. A Zika NS1-specific mAb (the irrelevant mAb) was used as a negative control. The concentration for 50% of maximal effect (EC50) was calculated using GraphPad Prism 8.
Affinity measurement
The KD measurement of isolated mAbs to SARS-CoV-2 S protein was conducted on the Octet K2 System (ForteBio, Fremont, CA) using the Bio-Layer Interferometry (BLI). Briefly, the Streptavidin (SA) sensor (Sartorius,18-5019) captured the biotin-labeled (EZ-LinkTM NHS-PEG12-Biotin, ThermoFisher Scientific, A35389) mAbs at 10 µg/ml, then serially diluted RBD protein was added. Three types of RBD proteins were measured for each Ab, including SARS-CoV-2 wild-type, XBB.1.16 (Vazyme, Nanjing, China), and SARS-CoV (ABclonal, Wuhan, China). After washing, the association and dissociation curves were obtained. The KD values were calculated using Data Analysis SPSS 11.0. A Zika NS1-specific mAb (the irrelated mAb) was used as a negative control.
Antibody competition for binding of ACE2 to RBD and S trimer
The competition assay was conducted on the Octet K2 System (ForteBio, Fremont, CA) using the Bio-Layer Interferometry (BLI). To measure antibody competition for binding of ACE2 to RBD, the Streptavidin (SA) sensor (Sartorius,18-5019) captured the biotin-labeled (EZ-LinkTM NHS-PEG12-Biotin, ThermoFisher Scientific, A35389) anti-D7 antibody at 40 µg/ml, which subsequently captured the SARS-CoV-2 RBD protein in the supernatant. After equilibrium, antibody (400 nM) and ACE2 protein (400 nM) or ACE2 protein and antibody were added in tandem. For control, we also determined the responses using antibody or ACE2 (400 nM) only. To measure antibody competition for binding of ACE2 to S trimer, the ACE2 (200 nM) was immobilized to Amine Reactive 2nd-generation (AR2G) biosensors (Sartorius, 18-5092) following the protocol recommended by the manufacturer. S trimer and antibody in four different ratios (1:0, 1:3, 1:15, 1:75) were premixed at room temperature for 10 min to ensure full reaction. The premixture was then presented to the biosensors loaded with ACE2. The mixture of buffer and S trimer was used for a control.
Competition ELISA for CYFN1006-1 or CYFN1006-2 and SA55
The competitive binding between mAbs of CYFN1006-1 or CYFN1006-2 and SA55 to SARS-CoV-2 RBD were measured using a capture ELISA (Supplementary Fig. 15). Briefly, the ELISA microplates precoated with anti-D7 tag Ab (Excellgen, EG-283, 4 μg/ml) were used to capture the SARS-CoV-2 RBD protein with D7-tag. Each mAb starting at 2 µg/ml was used for preparing a 5-fold serial dilution in the presence of 10 μg/ml competitor antibody and applied to the captured RBD ELISA plate. The binding curve of CYFN1006-1 or CYFN1006-2 was detected with a goat anti-human lambda IgG secondary antibody (Southernbiotech, 2070-05, 1 μg/ml), while that of SA55 was detected with a goat anti-human kappa IgG secondary antibody (Southernbiotech, 2060-05, 1 μg/ml). All samples were detected in duplicate. A normal human IgG purified from a pre-COVID-19 plasma sample was used as a negative competitor antibody control.
Production of pseudoviruses
Plasmids encoding the S of SARS-CoV, SARS-CoV-2 and its variants, as well as S from SARS-CoV and SARS-CoV-2 related sarbecoviruses, were synthesized. Pseudoviruses were generated by co-transfection of HEK-293T cells with the indicated S gene using PEI. Cells were cultured overnight at 37 °C with 5% CO2 and VSV-G pseudo-typed ΔG-luciferase (G*ΔG-luciferase, Kerafast) was used to infect the cells at a multiplicity of infection of 5 in DMEM with 10% fetal bovine serum (FBS), followed by washing the cells with PBS/2% three times and replaced culture medium by DMEM with 2% FBS. After 24 h, the transfection supernatant was collected and clarified by centrifugation at 4000 rpm for 10 min. Each viral stock was subsequently incubated with 20% I1 hybridoma (anti-VSV-G; CRL-2700, ATCC) supernatant for 1 h at 37 °C to neutralize the contaminating VSV-G pseudotyped ΔG-luciferase virus before titration and aliquoting for storage at −80 °C.
Pseudovirus neutralization assay
Neutralization assays were performed by incubating pseudoviruses with serial dilutions of monoclonal antibodies and assessing the reduction in luciferase gene expression, as previously described40. In brief, Vero-E6 cells were seeded in a 96-well plate at a concentration of 2 × 104 cells per well. The following day, pseudoviruses were incubated with serial dilutions of the specified monoclonal antibody in triplicate for 30 min at 37 °C. The mixture was then added to cultured cells and incubated for an additional 24 h. Subsequently, the luminescence was measured using the Luciferase Assay System (RG062M, Beyotime). IC50 was defined as the dilution at which the relative light units were reduced by 50% compared to the virus control wells (virus and cells), after subtraction of the background in the control groups with cells only. IC50 values were calculated using nonlinear regression in GraphPad Prism.
Viruses and biosafety
SARS-CoV GZ50 (GenBank accession number AY304495) was an archived clinical isolate at the Department of Microbiology, HKU. SARS-CoV-2 WT D614G (GISAID: EPL_ISL_497840), BA.1 (GISAID: EPI_ISL_6841980), XBB.1 (GISAID: EPI_ISL_15602393), EG.5.1 (GISAID: EPI_ISL_18461518), BA.2.86 (GISAID: EPI_ISL_18986956) and JN.1 (GISAID: EPL_ISL_18841631) strains were isolated from the respiratory tract specimens of laboratory-confirmed COVID-19 patients in Hong Kong. SARS-CoV-2 was cultured using Vero-E6-TMPRSS2. SARS-CoV was cultured in Vero-E6 cells. All the viruses were titrated by plaque assays. All experiments with infectious SARS-CoV-2 and SARS-CoV were performed according to the approved standard operating procedures of the Biosafety Level 3 facility at the Department of Microbiology, HKU41.
Authentic virus neutralization
An end-point dilution assay in a 96-well plate format was performed to measure the neutralization activity of select purified bsAbs as described previously27. In brief, each antibody was serially diluted (fivefold dilutions) starting at 50 μg/mL. Triplicates of each antibody dilution were incubated with the indicated live virus at an MOI of 0.1 in DMEM with 7.5% inactivated fetal calf serum for 1 h at 37 °C. After incubation, the virus–antibody mixture was transferred onto a monolayer of Vero-E6 cells grown overnight. The cells were incubated with the mixture for 70 h. The cytopathic effects were visually scored for each well in a blinded fashion by two independent observers. The results were then converted into percentage neutralization at a given concentration, and means ± SEM were plotted using a five-parameter dose–response curve in GraphPad Prism.
Hamster protection and Virus challenge
The use of all animals in the study was approved by The Committee on the Use of Live Animals in Teaching and Research (CULATR) of The University of Hong Kong (HKU). We have complied with all relevant ethical regulations for animal use. 6–8 weeks-old male golden Syrian hamsters were obtained from the Center for Comparative Medicine Research (CCMR) at HKU. The hamsters were housed in cages equipped with individual ventilation systems, maintained at a humidity level of 65%, and kept at an ambient temperature of 21–23 °C with a 12-h day-night cycle for proper care and management. Group sizes were chosen based on statistical power analysis and our prior experience in examining viral titers in SARS-CoV-2-infected hamsters or mice42,43.
To evaluate the antiviral effects of CYFN1006-1, hamsters were pre-treated for 8 h intraperitoneally with one dose of PBS or mAb CYFN1006-1 (20 mg/kg) before virus inoculation. All intranasal treatment in hamsters was performed under intraperitoneal ketamine (100 mg/kg) and xylazine (10 mg/kg) anesthesia. Virus-infected hamsters were euthanized at 4 dpi to collect NT and lung tissues for immunofluorescence staining, hematoxylin and eosin (H&E) staining, qRT-PCR, or TCID50 assays as we previously described44.
RNA extraction and quantitative reverse transcription polymerase chain reaction (qRT-PCR)
Viral RNA was extracted from homogenized animal tissues using RNeasy Mini Kit (74106, Qiagen). After RNA extraction, qRT-PCR was performed using QuantiNova Probe RT-PCR Kit (208354, Qiagen) or QuantiNova SYBR Green RT-PCR Kit (208154, Qiagen) with the LightCycler 480 Real-Time PCR System (Roche, Basel, Switzerland). The primer and probe sequences for SARS-CoV-2 E gene are: E_sgRNA_Forward, 5’-CGATCTCTTGTAGATCTGTTCTC-3’; E_sgRNA_Reverse, 5’-ATATTGCAGCAGTACGCACACA-3’; E_sgRNA_Probe, 5’-FAM-ACACTAGCCATCCTTACTGCGCTTCG-BBQ-3’. The primer sequences for hamster’s β-actin gene are: β-actin_Forward, 5’-ATGGCCAGGTCATCACCATTG-3’; β-actin_Reverse, 5’-CAGGAAGGAAGGCTGGAAAAG -3’.
Histology and Immunofluorescence staining
Infected hamster lungs were fixed by 4% paraformaldehyde for 24 h. Then, samples were washed with 70% ethanol, and a TP1020 Leica semienclosed benchtop tissue processor (Leica Biosystems, Buffalo Grove, IL, USA) was applied to embed lung samples in paraffin. Embedded samples were sectioned with a microtome (Thermo Fisher Scientific) and placed on microscope slides for drying at 37 °C overnight. Sample slides were dewaxed by washing in serially diluted xylene, ethanol, and double-distilled water. For antigens unmasking, diluted antigen unmasking solution (H-3300, Vector Laboratories) was heated until 85°C, then slides were put in solution and boiled for 90 s. Unmasked slides were stained with Sudan black B and blocked with 5% fetal bovine serum for 30 min. The in-house rabbit anti-SARS-CoV-2-N immune serum was applied onto samples and incubated at 4 °C overnight. Goat anti-rabbit immunoglobulin G(H + L) fluorescein isothiocyanate (65–6111), which functioned as a secondary antibody was purchased from Thermo Fisher Scientific. ProLong™ Diamond Antifade Mountant with DAPI (P36962, Thermo712 Fisher Scientific) was used to stain the cell nucleus while the slides were mounted. Images were taken with the Olympus BX53 fluorescence microscope (Olympus Life Science, Tokyo, Japan).
For H&E staining, tissue sections were stained with Gill’s hematoxylin and eosin. Images were acquired using the Olympus BX53 light microscope (Olympus Life Science, Japan). Five hamsters were sampled in each group and four to six sections from each animal were used for histology analysis.
Hamster lung and NT tissue infection virus titration
VeroE6-TMPRSS2 was obtained from the Japanese Collection of Research Bioresources (JCRB) Cell Bank. VeroE6-TMPRSS2 was cultured in Dulbecco’s modified Eagle’s medium (DMEM) (11965092, Gibco, USA) supplemented with 10% fetal bovine serum (FBS), 100 units penicillin, 100 ug/ml streptomycin, and 2% G418 (ant-gn-5, InvivoGen, China). To quantify the infectious titers of virus-infected hamster lung and NT tissues, a monolayer of VeroE6-TMPRSS2 cells in a 96-well plate was challenged with 10-fold serial diluted supernatants of homogenized tissues. After 96 h of incubation, the JN.1-induced cytopathic effect (CPE) was checked by visualization and calculated as we previously described45.
Protein expression and purification for Cryo-EM
The constructs used to express stable soluble Omicron XBB.1.16 (6 P) S and EG.5.1 (6 P) S were derived from our previous study27. Plasmid was transfected into HEK293F cells by polyethlenimine, supernatant was harvested after 72 h, and the S trimer is purified with the Ni-Sepharose resin from cytiva Reagents. The target protein was further purified by Superose 6 increase 10/300 column (GE Healthcare) in 20 mM Tris, 200 mM NaCl, pH 8.0 buffer solution.
Cryo-EM data collection and processing
The XBB.1.16 S trimer at 5 mg/mL was mixed with 5 mg/mL CYFN1006-1 at the molar ratio of 1:1.2 (S trimer/IgG), incubated at 4 °C for 30 mins, and the complex was separated on Superose 6 Increase 10/300 GL. The size and purity of the complex proteins were then further assessed by SDS-PAGE and diluted to 0.6 mg/mL in 20 mM Tris, pH 8.0, and 200 mM NaCl. The complex of EG.5.1 S and CYFN1006-2 was prepared by the above method. The 3 μL composite was added to a new light-emitting porous amorphous Ni–Ti alloy film supported by 400 mesh gold mesh. The sample was immersed in liquid ethane using Vitrobot IV (FEI, Thermo Fisher Scientific) and frozen for 2-s, -3blot force, 10-s waiting time.
Cryo-EM data for XBB.1.16-CYFN1006-1 and EG.5.1-CYFN1006-2 were collected automatically with EPU software on a 300 kV Titan Krios G4 microscope. The device is equipped with a Falcon 4i and Seletrics X-ray imaging filter (Thermo Fisher) with a slit width set to 20 eV. Collect EER movie stack in super-resolution mode, XBB.1.16-CYFN1006-1 nominal magnification of 130,000×, EG.5.1-CYFN1006-2 nominal magnification of 105,000×, corresponding physical pixel sizes of 0.932 Å and 1.19 Å. The doses were 1080 frames and 1737 frames, respectively46. The defocusing range is −1.0 to −3.0 μm, and the total dose is about 50e−/Å2. Data processing is performed via modules on RELION v3.1 or cryoSPARC v4.2.1. All micrographs were imported into cryoSPARC for patch CTF estimation47, particle pickup, and 2D classification. Then, select the good particles and import them into Relion for 3D automatic thinning, 3D thinning, and 3D classification. The resolution of the report is based on the gold standard Fourier Shell correlation (FSC) of 0.143. The data processing flow is summarized in Supplementary Figs. 5 and 9.
Model building and refinement
EMready48 is used to sharpen maps for model building. SWISS-MODEL49 is used to generate initial models. The models were fitted into maps using UCSF Chimera50, manually built in COOT51, followed by multiple rounds of real space refinement in PHENIX52. Model validation using PHENIX. Figures were prepared using UCSF Chimera and UCSF ChimeraX53. Statistics of data collection and model refinement are shown in Table 1.
Statistics and reproducibility
Data on figures represented means and standard error of the mean. Comparison between the two groups was performed with two-sided Student’s t tests. Differences were considered statistically significant when p < 0.05. Data analysis was performed with GraphPad Prism 8.0.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Responses