Pulling back the mitochondria’s iron curtain
Introduction
Iron played a critical role in the emergence and establishment of life on earth. In the metal-rich anaerobic oceans where early life evolved1, iron’s wide redox potential likely enabled rudimentary metabolism, electron transport, and energy transfer2,3. Shaped by these origins, modern lifeforms also heavily rely on iron cofactors to catalyze fundamental reactions. Conservative estimates indicate that hundreds of human proteins bind iron4, facilitating DNA replication and repair, gene expression, oxygen transport, and beyond. These proteins either engage an iron ion directly or bind to an iron complex as an iron-sulfur (Fe-S) cluster or heme. While these iron cofactors are tightly “tuned” by their binding proteins to create selective reduction potential windows tailored to their biological functions, collectively, they span a remarkable range from −770 to +440 mV5. Being so customizable, these cofactors enable a wide spectrum of redox reactions.
Within the eukaryotic cell, the mitochondrion stands out both for its notable contributions to iron cofactor biosynthesis and utilization. First, both heme and Fe-S cluster biosynthesis occurs, in part, in mitochondria. Comparative evolutionary analysis indicates that the latter is among the most conserved mitochondrial-residing pathways6. Second, the relative engagement of iron cofactors within the mitochondrial proteome is ~3.5-fold higher than the total proteome4. These cofactors are prominent in the electron transport chain, mitochondrial anabolic and catabolic pathways, and mitochondrial translation apparatus. In addition, mitochondria can also take part in iron storage, by virtue of mitochondrial ferritin (FTMT). Thus, mitochondria are at the crossroads of cellular iron metabolism and engagement. Several excellent recent reviews have discussed how cells acquire iron, and how this metal is subsequently transferred to the mitochondrial matrix7,8. Here we will highlight how iron is metabolized or utilized in the mitochondria to support the biology of this multifaceted organelle (Fig. 1).
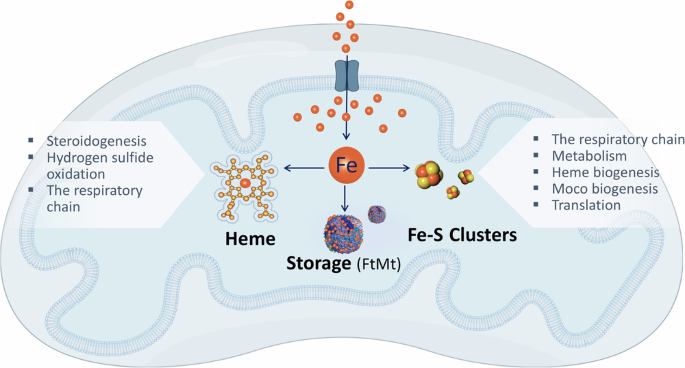
Upon entering the mitochondrial matrix, iron can be channeled to three pathways: heme biosynthesis, iron-sulfur (Fe-S) cluster biogenesis, or storage in mitochondrial ferritin (FtMt). The cofactors produced by mitochondrial iron, namely Fe-S clusters or heme, subsequently support various essential mitochondrial tasks.
Mitochondrial iron handling
Iron-sulfur cluster biogenesis
Made up of different combinations of inorganic iron and sulfur, Fe-S clusters can exist in numerous forms, the most common being [2Fe-2S], [3Fe-4S], and [4Fe-4S] clusters9. As electrons are delocalized over both iron and sulfur ions in the cluster10, Fe-S clusters are highly suited for electron transfer, redox sensing, or catalysis. In addition, these versatile cofactors can also take part in sulfur donation reactions and have been shown to contribute to the structural stability of the proteins into which they are integrated.
While Fe-S cluster formation was originally thought to occur spontaneously within cells11,12,13, it is now appreciated that clusters must be actively synthesized and relayed to the proteins that require them14,15,16 (Fig. 2 left). The first step of biogenesis involves the iron-sulfur cluster (ISC) machinery, which catalyzes the synthesis of a [2Fe-2S] cluster from ferrous iron, sulfur (provided by cysteine), and electrons. The cluster is built upon the scaffold protein, ISCU, and relies upon the rate-limiting activity of the cysteine desulfurase complex NFS1-LYRM4-NDUFAB1. Cluster biogenesis also demands electrons, provided by ferredoxin 2 (FDX2), and is allosterically activated by frataxin (FXN). Once the initial [2Fe-2S] cluster has been generated, it is transferred to the glutaredoxin 5 (GLRX5) dimer with the help of the chaperone/co-chaperone pair HSPA9/HSC20. This process requires energy supplied by ATP hydrolysis carried out by HSPA917. At this step, an intermediate is generated that can be relayed to the cytosol via the inner mitochondrial membrane (IMM) transporter ABCB7, supporting the formation of cytosolic and nuclear Fe-S clusters. Of note, several reports have indicated that the ISC pathway may be simultaneously active in the cytosol18,19,20. Finally, the generation of mitochondrial [4Fe-4S] clusters is carried out by ISCA1 and ISCA2, which reductively couple two [2Fe-2S] clusters10,21. This reaction also requires IBA57, whose mechanistic role remains poorly defined, and electron transfer from FDX2. Together, this complex multistep pathway ensures the controlled production and relay of labile clusters to their various acceptor proteins.
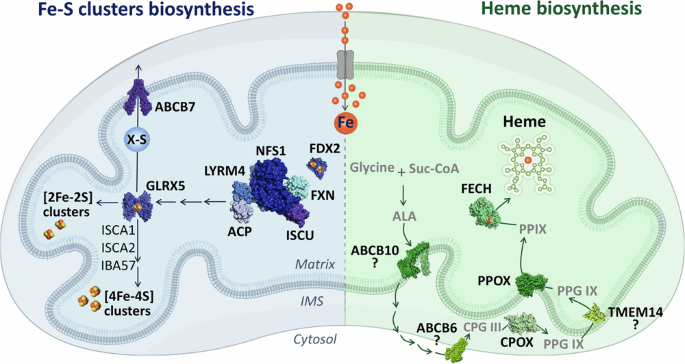
Mitochondrial iron-sulfur (Fe-S) cluster biogenesis (left) is initiated by a protein complex, comprised of key proteins such as the ISCU scaffold, the cysteine desulfurase NFS1, and the allosteric activator FXN. This complex generates 2Fe-2S clusters, which are relayed to GLRX5. Subsequent machinery, comprising ISCA1, ISCA2, and IBA57, can build on these clusters to form 4Fe-4S clusters. Alternatively, some intermediate can be handed off to the cytosol via the inner membrane transporter ABCB7. Concurrently, heme biosynthesis (right) begins with the condensation of glycine and succinyl-CoA (Suc-CoA) to form 5-aminolevulinic acid (ALA). ALA is exported to the cytosol, potentially by ABCB10, where it undergoes further processing before being re-imported into the mitochondrial inner membrane space, possibly via ABCB6. Within this compartment, CPOX catalyzes the formation of protoporphyrin IX (PPIX), which is then transported to the mitochondrial matrix and converted to heme by FECH. PDB IDs for protein structures: ABCB7 (7VGF), GLRX5 (2WUL), FDX2 (2Y5C), LTRM4-NFS1-FXN-ISCU-ACP (8PK8), ABCB10 (7Y49), FECH (1HRK), PPOX (3NKS), CPOX (2AEX), ABCB6 (7DNY), and TMEM14 (2LOR).
Heme biosynthesis
Heme, an iron-containing tetrapyrrole22, can unlock diverse abilities including oxygen binding23, electron transport24, and enzymatic catalysis. Thus, heme is essential for virtually all aerobic lifeforms. Unlike the case for Fe-S clusters, cells can take up heme from their environment. However, the relative contributions of endogenous vs. exogenous supply to organellar and cellular heme pools remains an area of active investigation.
Heme biosynthesis in most metazoans involves a coordinated sequence of eight enzymatic reactions, which traverse the mitochondria and cytosol (Fig. 2 right). A notable exception to this biosynthetic capability are the nematode Caenorhabditis elegans and other parasitic helminths, which are heme auxotrophs25. However, the majority of animals are capable of synthesizing heme de novo from basic substrates: ferrous iron, glycine, and succinyl-CoA. The mitochondrial enzyme ALA synthase (ALAS) initiates the rate-limiting condensation of succinyl-coenzyme A and glycine to form 5-aminolevulinate (ALA)26. Once ALA is generated, it must be exported to the cytosol via an unidentified transporter, although there are indications that ABCB10 might be involved in this process27,28. In the cytosol, ALA is converted into coproporphyrinogen III (CPG III) through a series of four reactions. The final steps of heme biosynthesis occur back in mitochondria. First, CPG III is transported into the intermembrane space, possibly by ABCB629, where it is oxidized to protoporphyrinogen IX (PPG IX) by coproporphyrinogen III oxidase (CPOX). This is notable, as there are very few metabolic reactions that are carried out in the intermembrane space. Next, PPG IX is transported into the mitochondrial matrix, potentially via the transmembrane protein TMEM14C30, and oxidized to form protoporphyrin IX (PPIX) by the IMM enzyme protoporphyrinogen oxidase (PPOX)22. The final mitochondrial step involves the insertion of ferrous iron into the protoporphyrin ring to form the heme group by ferrochelatase (FECH)31. Thus, the complex heme biosynthesis pathway both starts and ends in mitochondria, although the benefits conferred by this elaborate spatial segregation remain unclear.
Mitochondrial iron sequestration
Mitochondria from varied sources have been shown to contain a sizeable 20–50% of the total cellular iron content32,33. Excess iron can be stored in the mitochondrial form of ferritin (FTMT), a homopolymer that is structurally and functionally similar to the cytosolic ferritin heavy chain34. By sequestering free iron, FTMT may limit the iron-driven production of reactive hydroxyl radicals via the Fenton reaction35. Notably, FTMT knockout mice are viable and display no significant defects in systemic iron metabolism under baseline conditions36, but have been shown to be susceptible to oxidative or neurotoxic insults37,38,39,40. Further hinting at a role in oxidative regulation, FTMT is primarily expressed in the testis41, rather than iron-rich tissues such as the liver and spleen. Taken together, these data suggest that the primary function of FTMT may be regulatory and protective, rather than a constant buffer of mitochondrial iron pools42,43.
Iron-dependent mitochondrial pathways
Fe-S cluster-dependent processes
The respiratory chain
The mitochondrial respiratory chain is a central redox hub of the eukaryotic cell. While classically depicted as four macromolecular IMM complexes working in a linear fashion to couple electron transfer with proton pumping, the respiratory chain is, in fact, a dynamic sink for many cellular redox reactions. In metazoans, these include de novo pyrimidine biosynthesis, sulfide metabolism, fatty-acid oxidation, amino acid metabolism, and glycolysis.
Consequently, it is not surprising that the respiratory chain is rich in Fe-S clusters that help channel electrons (Fig. 3A)44,45. Specifically, the ‘early’ steps of the respiratory chain that feed electrons to coenzyme Q (CoQ)- Complexes I and II- are abundant in Fe-S clusters. Complex I (NADH: ubiquinone oxidoreductase) is the primary entry point of electrons into the respiratory chain, passing reducing equivalents from NADH to CoQ46. To carry out this reductive reaction, Complex I binds flavin and, depending on the organism, up to ten Fe-S clusters47. Working as an “electron wire”, these Fe-S clusters connect the two substrate-binding sites that lay over 90 Ȧ apart48,49. Complex II (succinate dehydrogenase) works at the interface of the tricarboxylic acid (TCA) cycle and the respiratory chain. While not contributing to proton pumping, complex II oxidizes succinate to fumarate and transfers these electrons to CoQ. Three Fe-S clusters, all bound to the subunit SDHB, are essential for this activity50. Lastly, complex III (cytochrome bc1) accepts electrons relayed via CoQH2 and relays them to cytochrome c. A [2Fe-2S] cluster in this complex is coordinated by the Rieske iron–sulfur protein (UQCRFS1), and directly accepts electrons from CoQH2. This represents the first and rate-limiting step of the Q cycle, in which these electrons will be subsequently relayed to cytochrome c (i.e. the high-potential redox chain). In parallel, electrons produced via fatty-acid oxidation and amino acid catabolism are also channeled to CoQ via an Fe-S cluster-binding protein. Specifically, the [4Fe-4S] cluster-binding electron transfer flavoprotein dehydrogenase (ETFDH) aids in transferring electrons from multiple mitochondrial acyl-CoA dehydrogenases to CoQ51. In this way, multiple reductive sources “feed” electrons to the respiratory chain via Fe-S clusters.
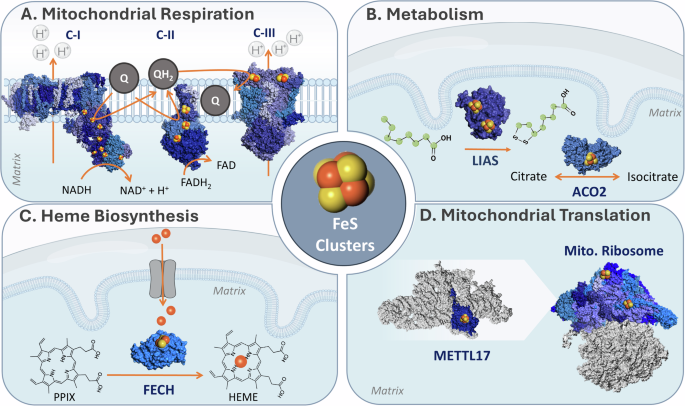
A Mitochondrial respiration. Fe-S clusters take part in electron transfer in complexes I, II, and III in the respiratory chain, found in the inner mitochondrial membrane. B Mitochondrial metabolism. (i) Lipoic acid synthesis requires LIAS, which catalyzes the insertion of two sulfur atoms into a protein-bound octanoyl chain; (ii) The TCA enzyme mitochondrial aconitase, which reversibly converts citrate to isocitrate. C Heme biosynthesis. Fe-S clusters are required in the second and final steps of heme biosynthesis. The last is present in the mitochondria, whereby FECH catalyzes the insertion of a ferrous iron into the protoporphyrin ring. D Mitochondrial translation. The biogenesis and structure of the mitochondrial ribosome require Fe-S clusters. The late-stage small mitoribosome subunit assembly factor, METTL17, binds an 4Fe-4S cluster essential for its stability. In addition, two Fe-S clusters are found in the small subunit of the mitoribosome, where they stabilize protein-protein interaction interfaces. PDB IDs for protein structures: Complex I(4WZE), Complex II (8GS8), Complex III (5XTE), LIAS (5EXK), ACO2 (5ACN), FECH (1HRK), METTL17 (8OM2), and Mitoribosome (6QZP and 7P2E).
Mutations in the mitochondrial Fe-S cluster biosynthesis or relay machineries have been shown to dramatically blunt respiration in multiple model systems, most notably affecting complex I and II stability. These clusters are also labile in the face of environmental insults, such as oxygen toxicity52. Recent work has highlighted an auxiliary Fe-S binding protein which may buffer complex I Fe-S cluster levels- the CDGSH iron-sulfur domain 3 protein (CISD3 or MiNT). Found in the mitochondrial matrix, CISD3 belongs to the highly conserved [2Fe-2S]-binding NEET protein family. Fe-S clusters bound to NEET proteins can participate in electron transfer reactions53,54, but are bound by a labile and redox-active binding pocket. This later trait supports cluster transfer between NEET and acceptor proteins. CISD3 has been shown to engage with and donate its Fe-S cluster to Complex I subunit NDUFV255. Interestingly, NDUFV2 binds the N1a cluster in complex I, which is not part of the “electron wire”. Rather, the N1a cluster has been suggested to prevent excessive ROS formation or to be required for Complex I assembly and stability.
In summary, Fe-S cluster are crucial for mitochondrial respiration, enabling ATP synthesis, redox rebalancing (NADH, CoQ), and the mitochondrial membrane potential that drives transport of proteins and metabolites across the MIM.
Metabolism
Fe-S clusters are woven throughout various metabolic mitochondrial pathways (Fig. 3B), affecting this organelle’s capacity for anabolism and catabolism in distinct ways. While some of these clusters take part in electron transfer reactions, others enable alternative functions such as catalysis, regulation, and sulfur donation.
TCA cycle and its cofactors
The TCA cycle, a core axis of mitochondrial metabolism, relies on Fe-S clusters both directly and indirectly at several different nodes (Fig. 3B). First, the TCA enzyme mitochondrial aconitase (ACO2), which catalyzes the reversible isomerization of citrate to isocitrate56, has a [4Fe-4S] cluster at its substrate-binding site. ACO2 exemplifies the catalytic capacity of clusters- this [4Fe-4S] cluster acts as a Lewis acid, by virtue of a Fe ion in the cluster that is not bound by ACO257. Second, Complex II, highlighted in the section “The respiratory chain”, converts succinate to fumarate through the action of its three Fe-S clusters. Finally, two key dehydrogenases associated with the TCA cycle require a cofactor generated via an Fe-S cluster-dependent reaction. Specifically, the catalytic activities of pyruvate dehydrogenase and α-ketoglutarate dehydrogenase are critically dependent on the production of lipoic acid, a straight-chain fatty-acid containing two sulfhydryl groups. The biogenesis of this cofactor necessitates the involvement of two Fe-S cluster-binding proteins: lipoic acid synthase (LIAS) and ferredoxin 1 (FDX1). LIAS binds two [4Fe-4S] clusters—the first mediates a radical SAM reaction that activates the aliphatic chain for sulfur attachment, while the second acts as a sulfur donor and must be replenished after each reaction58. However, the source of the electron that jumpstarts the radical SAM reaction on LIAS has remained unclear for some time. Several groups have recently shown that FDX1, apart from its previously established roles in processes such as steroidogenesis57, is also vital for lipoic acid biosynthesis59,60,61. By contributing electrons for the radical SAM reaction, FDX1 plays a crucial role in maintaining the TCA cycle. Altogether, by enabling the TCA cycle, Fe-S clusters take part in the controlled combustion of nutrients and supply metabolic precursors for nucleotide, lipid, and protein biogenesis.
Mitochondrial glutathione balance
The small peptide glutathione plays a principal role in cellular antioxidant defense. Readily transitioning between its reduced monomeric and oxidized dimeric forms (GSH vs. GSSG), glutathione can neutralize reactive oxygen species both directly and indirectly (via antioxidant enzymes). This buffering capacity is particularly pertinent in the mitochondria, where the respiratory chain can produce the highly reactive O2.– oxidant62. However, glutathione is generated in the cytosol, and while had long been known that up to 15% of cellular glutathione is found in the mitochondria63, how it passes the IMM remained unresolved until late. Two independent studies revealed that GSH is transported into the mitochondrial matrix via the solute carrier family 25 paralogues- SLC25A39 and SLC25A4064,65. While SLC25A40 appears to be constitutively expressed, SLC25A39 protein is strongly upregulated upon GSH depletion. How is this autoregulation mechanism achieved? Further work into SLC25A39 has revealed that it binds an [2Fe-2S] cluster within a cytosolic loop. Importantly, this cluster can be competed out of SLC25A39 with GSH, destabilizing the protein under high GSH conditions66,67. Thus, Fe-S cluster binding in SLC25A39 highlights the metabolite-sensing potential of these cofactors.
Heme biogenesis
Heme and Fe-S clusters are tightly linked- the biosynthesis of the former requires the latter. Specifically, both the second68 and last69 steps of heme biosynthesis are catalyzed by Fe-S cluster proteins. Focusing on the terminal mitochondrial reaction, it has been shown that FECH engages a [2Fe-2S] cluster (Fig. 3C). This cluster does not appear to have a catalytic role, but rather stabilizes FECH’s tertiary and quaternary structure, enabling the formation of the dimer interface70. Indeed, FECH protein is downregulated in several models of Fe-S cluster depletion71,72,73. Whether the Fe-S cluster on FECH has additional functions remains to be explored.
Molybdenum cofactor (Moco)
Molybdenum is a vital trace element for animals and is essential for human development. Four key redox enzymes engage in molybdenum-based catalysis in humans: sulfite oxidase, xanthine dehydrogenase, aldehyde oxidase, and mitochondrial amidoxime reductase74. However, on its own, molybdenum is biologically inactive, and must be complexed by a scaffold to unlock its biocatalytic potential. The molybdenum cofactor (Moco) is a pterin-based scaffold present in all kingdoms of life, in which molybdenum is coordinated by sulfur. Moco biosynthesis requires the coordinated actions of four human genes (MOCS1, MOCS2, MOCS3, and GEPH sequentially75) encoding six proteins that localize to the mitochondria and cytosol. Fe-S clusters play a crucial role in this process76, as the mitochondrial MOCS1A enzyme, which coordinates the first step in Moco biosynthesis, binds two [4Fe-4S] clusters per monomer77. One cluster has a well-established role in mediating a radical SAM reaction. However, the function of MOCS1A’s auxiliary cluster remains unclear, possibly taking part in substrate coordination. Taken together, Fe-S cluster biosynthesis controls the primary step of Moco production and the activity of Moco-containing enzymes in animals. One exception to this rule was recently discovered in nematodes, when it was demonstrated that C. elegans (apart from producing Moco) can import significant amounts of Moco from the bacteria on which it feeds78,79. This raises intriguing questions as to whether Moco uptake occurs in other animals, and if any regulatory crosstalk exists between Fe-S clusters depletion and environmental Moco acquisition.
Translation
As the respiratory chain is dually encoded by the nuclear and mitochondrial genomes, protein synthesis by matrix-residing mitoribosomes is essential to support mitochondrial bioenergetics80. Recent discoveries have uncovered the importance of Fe-S clusters in the mitochondrial translation process (Fig. 3D). Both the mitoribosome assembly factor methyltransferase like protein 17 (METTL17) and the mitoribosomal small subunit (mt-SSU) contain Fe-S clusters. METTL17, a highly conserved protein localized to the mitochondrial matrix81, acts as a late-stage assembly factor for the mt-SSU. Biophysical and cryo-EM studies have revealed that METTL17 contains a highly labile [4Fe-4S] cluster- essential for its interaction with the mt-SSU and overall structural stabilization73,82. Thus, METTL17 acts as an “Fe-S cluster checkpoint” that matches mitoribosome assembly with Fe-S cluster availability. In addition, structural and functional studies have revealed that the mt-SSU includes two [2Fe-2S] clusters82,83,84. In a rather unique manner, both clusters are simultaneously bound by two different proteins- one engaging with bS18m and bS6m and the other by mS25 and bS16m. Comparative analysis between the mitoribosome and a bacterial homolog highlighted that these 2Fe-2S clusters are found where bacterial ribosomal RNA (rRNA) is typically engaged. This led the authors to the conclusion that these clusters facilitate mt-SSU complex formation, contributing to the stability of newly added proteins in regions where the rRNA has been deleted over the course of evolution.
Mitochondrial hemoproteins and pathways
Steroidogenesis
Mitochondria are crucial sites for steroid hormone biosynthesis85 (Fig. 4A). Steroidogenesis requires the mobilization of cholesterol to the IMM, where it is subsequently converted into various steroids through a series of reactions involving cytochrome P450 and hydroxysteroid dehydrogenase enzymes86,87. Mitochondrial cytochrome P450 enzymes, embedded in the IMM with most of their protein body facing the matrix, bind a single heme moiety88,89. By virtue of their redox center, these enzymes function as monooxygenases, activating molecular oxygen using electrons donated by nicotinamide adenine dinucleotide phosphate (NADPH). For example, the first and rate-limiting step in steroidogenesis is the conversion of cholesterol to pregnenolone by a single mitochondrial P450 enzyme, cholesterol side-chain cleavage enzyme (P450scc or CYP11A1)90. Thus, P450scc activity serves as a key regulator in the biogenesis of these crucial signaling molecules. Pregnenolone serves as the precursor for multiple different classes of steroid hormones, through divergent reactions that occur in both the mitochondria and ER. Of note, the terminal steps in the biosynthesis of both glucocorticoids and mineralocorticoids occur in the mitochondria. These are mediated by 11β-hydroxylase (P450c11β or CYP11B1), which generates cortisol (followed by activities of the ER enzymes P450c21 and P450c17), and aldosterone synthase (P450c11AS or CYP11B2) which produces aldosterone91,92,93,94. Cytochrome P450 enzymes cannot oxidize NADPH directly, instead these electrons are relayed via the Fe-S cluster containing protein FDX189,95. In this manner, both iron-based cofactors are essential for steroidogenesis.
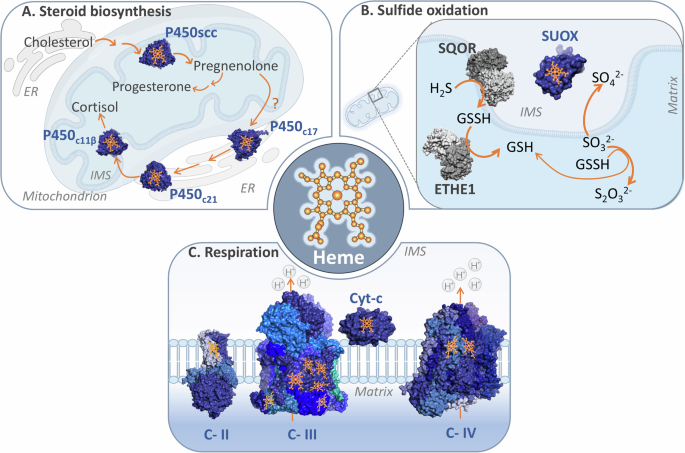
A Mitochondrial steroid biosynthesis, schematic review depicts the mitochondrial steroidogenesis, involving the transport of cholesterol to the IMM by the StAR protein. Cholesterol is then converted to pregnenolone by the enzyme P450scc through three monooxygenase reactions. Pregnenolone then serves as a precursor for various steroid synthesis, including progesterone and cortisol, with further modifications occurring in the endoplasmic reticulum (ER) and mitochondria. The process relies on the action of the cytochrome P450 family enzymes and the transfer of electrons from NADPH via FDX1; B The mitochondrial sulfide oxidation pathway, schematic of the canonical sulfide oxidation pathway in mitochondria, illustrating the reduction of H2S to GSSH by SQOR. GSSH is further oxidized by ETHE1 to GSH, to finally produce GSH and S2O32-. The SO32- metabolite, is alternatively oxidized to SO42- by SUOX; C Heme groups’ role of mitochondrial respiration complexes, schematic representation of the electron transfer process within the mitochondrial electron transport chain. Electrons are transferred from Complex II to Complex IV via cytochrome c. The electron shuttles between all enzymes, are transferred to the heme groups fixed at their structure. PDB IDs for protein structures: P450SCC(3MZS), P450C21(4Y8W), P450C11β(6M7X), P450C17(6WW0), SUOX(1MJ4), SQOR(6OI5), ETHE1(4CHL), Complex II(8GS8), Complex III(8UGD), Complex IV(5Z62), and Cyt-c(2N9J).
Hydrogen sulfide oxidation
Hydrogen sulfide (H2S) is a two-faced molecule. It can act as a toxin, primarily through its ability to engage heme in Complex IV in the respiratory chain96, blocking its activity. Conversely, H2S also constitutes a signaling molecule, as revealed by a landmark paper showing its neuromodulatory effects97. Thus, its levels must be tightly controlled. The process of H2S clearance takes place in the mitochondria (Fig. 4B). The terminal step in this pathway requires a heme- and molybdenum-dependent enzyme named sulfite oxidase (SUOX). In the inner membrane space, SUOX oxidizes sulfite (so32−) to sulfate (so42−)98, which can then be excreted in the urine. Electrons from the H2S oxidation contribute to mitochondrial respiration, as SQOR donates its reducing equivalents to coenzyme Q99. However, disease-associated SUOX mutations have revealed that heme binding may not be strictly required for SUOX activity100. Moreover, H2S can also be oxidized by additional proteins, among them heme-containing cytochrome c101. This alternate mode of clearance may be relevant in cells and tissues with low levels of the H2S oxidation pathway. It is thus notable that H2S both inhibits and contributes to respiratory chain activity, suggesting that it induces alternative patterns of electron flow in the respiratory chain102.
The respiratory chain
Heme is crucial for mitochondrial respiration, aiding in both electron transfer and structural stability in complexes II, III, and IV (Fig. 4C).
One heme moiety is situated between the two transmembrane proteins of complex II, specifically SDHC and SDHD. Initially, it was hypothesized that this heme, along with the Fe-S clusters, would be part of the redox chain that mediates electron transfer from succinate to CoQ. However, subsequent structural studies revealed that the distances between the redox centers make it unfavorable for heme to be involved in electron transfer103. Therefore, this heme is now considered to be primarily structural104, although it has been speculated that it may play additional regulatory roles.
Complex III contains several heme moieties that form two alternate redox chains, generating the bifurcating Q cycle105. In this manner, the oxidation of one CoQH2 molecule is coupled with the reduction of cytochrome c and CoQ at opposing sides of the complex. The high-potential redox chain, discussed in section “The respiratory chain”, is made up of one heme on cytochrome c1 and the Fe-S cluster engaged by UQCRFS1. As previously mentioned, this chain passes the first electron from CoQH2 to cytochrome c. The low-potential redox chain is composed of two hemes bound to cytochrome b. It transfers the second electron from the CoQH• anion to another CoQ106,107. The cycle then repeats. How is this bifurcation enabled, or in other words, why don’t both electrons follow the same path? It’s been shown that once the Fe-S cluster on UQCRFS1 is reduced, this protein undergoes a dramatic conformational change108,109,110. This distances the [2Fe-2S] cluster from the CoQH• anion, making electron transfer more favorable through the low-potential redox chain.
Cytochrome c, a soluble protein localized on the IMM, contains a single heme group. By shuttling electrons one at a time from complexes III to IV, it serves as a crucial linker in the respiratory chain111. The heme moiety in cytochrome c is covalently attached to the protein via a conserved CXXCH motif by the enzyme holocytochrome c synthase (HCCS). It is thought that such covalent linkages provide stability, making cytochrome c less likely to lose its cofactor112. In the mature protein, one edge of the heme group is positioned ~5 Ȧ from the surface of the protein113,114, resulting in small edge-to-edge distances with the redox centers of complex III and IV.
Upon reaching complex IV (cytochrome c oxidase), cytochrome c transfers its electron to a bi-copper center located in the mitochondrially encoded MT-COX2 subunit115. This electron is subsequently channeled through three redox centers in MT-COX1. This core subunit is membrane-bound and contributes to both proton pumping and oxygen reduction reactions. To this end, MT-COX1 binds two heme groups and an additional copper ion. A binuclear redox center, made of one heme and copper found ~5 Ȧ apart116,117,118, is the site of molecular oxygen binding and reduction to water. This process is coupled to the flow of four protons across the IMM119 through a hydrated channel in MT-COX1120. In this manner, heme molecules are central for the terminal step of the respiratory chain, metabolizing ~90% of molecular oxygen found within the cell.
Mitochondrial iron-binding proteins
Notably, a handful of mitochondrial proteins engage iron directly.
Mitochondrial 5-demethoxyubiquinone hydroxylase (COQ7), a soluble protein found on the matrix side of the IMM, is a conserved enzyme essential for ubiquinone biosynthesis. Part of a multiprotein CoQ metabolon121, COQ7 bears a di-iron center that catalyzes penultimate step of ubiquinone biosynthesis122,123,124,125. Biochemical analysis has revealed that NADH can directly serve as a reductant for COQ7, with no requirement for an additional reductase protein component.
The ethylmalonic encephalopathy 1 protein (ETHE1) is an iron-coordinating metalloprotein that takes part in sulfide catabolism126,127. Localized in the mitochondrial matrix, ETHE1 oxidizes glutathione persulfide to sulfite (SO32−) while consuming molecular oxygen and water128. The mononuclear iron in ETHE1 likely takes part in catalysis, being found at the active site and near the product127,129.
Signaling mitochondrial iron depletion
Attenuated mitochondrial iron stores trigger several cellular homeostasis signaling cascades. Collectively, these counterbalancing measures act to elevate iron availability, while simultaneously blunting global protein translation and activating a distinct transcriptional stress response program. In this manner, such signaling responses enable cells to survive acute phases of iron depletion.
Iron starvation response and ferritinophagy
Cells have evolved sophisticated sensing pathways to detect and rectify iron deficiencies130. The absence of this element may arise from either a scarcity of environmental iron or a failure in uptake, as in the case of lysosomal dysfunction131. Changes in its typically acidic pH leads to iron retention within the lysosome, resulting in impaired mitochondrial function132.
In metazoan cells, iron homeostatic mechanisms include post-transcriptional response pathways and targeted release of intracellular iron stores. Intriguingly, both regulatory pathways are not responsive to iron directly, but instead react to Fe-S cluster levels133.
First, post-transcriptional control of iron levels is carried out by the iron-regulatory proteins, IRP1 and 2134. Together, these orthologous proteins regulate the translation and stability of mRNAs bearing an iron response element. This mRNA motif is found in the untranslated regions of key iron metabolism transcripts, capable of modulating cellular iron uptake and storage135. How are IRP1/2 activated during iron deficiency? IRP1 binds a [4Fe-4S] cluster at its mRNA binding pocket, so that it can only engage the iron-responsive elements in the absence of this cofactor136. Conversely, IRP2 is degraded by a [2Fe-2S] cluster-binding E3 ubiquitin ligase137, so that its protein levels are significantly elevated during iron scarcity. IRP2 has also been shown to sense Fe-S cluster levels regardless of its degradation138, through a yet-unclear mechanism.
Second, excess intracellular iron is sequestered by cytosolic ferritin, and can be dynamically released upon iron depletion via ‘ferritinophagy’139,140. This selective autophagic pathway targets ferritin to the lysosome through the actions of the nuclear receptor coactivator 4 (NCOA4)141,142. It was recently discovered that NCOA4 engages a [3Fe-4S] cluster143,144. In the absence of its Fe-S cluster, NCOA4 is stabilized and can engage with ferritin. These two mechanisms are in line with the iron accumulation observed in various tissue samples of Fe-S cluster deficiency patients. In this manner, Fe-S clusters serve as pivotal gauges of cellular iron stores.
Integrated stress response
Studies in various models of mitochondrial dysfunction have consistently identified an activation of the integrated stress response (ISR). This pathway involves the phosphorylation of eukaryotic translation initiation factor 2 alpha (eIF2α) by one of the four eIF2α kinases, reducing global protein synthesis and activating transcription factor ATF4 to restore homeostasis145,146. Each of these four kinases is triggered by a specific insult147, such as amino acid deficiency148,149, unfolded protein stress150, heme depletion151, and viral infection152,153.
Until recently, it was not clear how mitochondrial dysfunction triggers the ISR. There was evidence that the secondary depletion of amino acids caused by blocked respiration might be the primary cause of ISR activation154. However, this phenomenon was observed in some contexts, but not others, hinting that alternate ISR activation pathways may exist. Breakthrough work in 2020 showed that it is, in fact, heme-regulated eIF2α kinase (HRI) that is responsive to mitochondrial stress155,156. Classically, HRI has been studied in the context of heme depletion. However, under mitochondrial stress, HRI activation occurs through a distinct mechanism, which centers on the mitochondrial “signal relay” protein DELE1. Mitochondrial stress stimulates the cleavage of DELE1 by the protease OMA1, resulting in the accumulation of the C-terminal domain of DELE1 in the cytosol. This short form of DELE1 then binds and activates HRI, initiating the long-observed ISR transcriptional response.
A recent study by the Sekine group demonstrated that under iron deficiency, the HRI-ISR cascade is also activated157. Surprisingly, this is not due to heme-induced activation of HRI, but rather triggered by DELE1, although there are some key differences with the established model. In this context, DELE1 is not cleaved by OMA1, but rather is stabilized on mitochondria due to mitochondrial import arrest157. This stabilization results in the exposure of DELE1’s C-terminal domain to the cytosol, where it interacts with and activates the HRI-ISR pathway. The Chan group have further extended these findings to show that HRI (and phospho- eIF2α) recruitment to the mitochondrial outer surface also triggers autophagy158. Taken together, it appears that mitochondria have co-opted the ISR as a local iron monitoring system, which can mark and eliminate organelles critically depleted for this essential metal.
OMA1-DELE1-dependent ISR signaling may also protect against ferroptosis159, an iron-dependent form of cell death160 that is further discussed in the section “Mitochondrial iron at the crossroads with human diseases”. Recent work has demonstrated that early-onset cardiomyopathy caused by respiratory chain dysfunction activates the ISR signaling cascade. This response acts to delay pathology and suppresses ferroptosis, by promoting antioxidative factors, such as glutathione159. This raises intriguing questions as to the protective role of the ISR in other contexts of iron overload.
Mitochondrial iron at the crossroads with human diseases
Given the diverse and vital roles of mitochondrial iron, it is not surprising that imbalances can have pathological consequences. These span the range from severe but rare genetically encoded disorders to more common diseases161,162. Here, we highlight human pathologies that result from genetic or environmental alterations in mitochondrial iron handling pathways.
Fe-S cluster deficiency
Mutations in genes related to Fe-S cluster biogenesis or transfer can cause various pathologies. The most prominent Fe-S cluster disorder is Friedreich’s ataxia163, which affects 1:50,000 individuals. This autosomal recessive disease is caused by mutations in the nuclear gene FXN164,165, which encodes for an allosteric activator of Fe-S cluster biosynthesis in the mitochondria. While the hallmark of this disease is ataxia, patients are also prone to diabetes, scoliosis, hearing and vision loss, and cardiomyopathy166,167,168,169. The latter is the leading cause of premature mortality in Friedreich’s ataxia170. In 2023, Omaveloxolone received FDA clearance for Friedreich’s ataxia171, making it the first drug approved for any monogenic mitochondrial disease172. Omavaloxolone engages the cell’s endogenous antioxidant response, and while it can attenuate the progression of Friedreich’s ataxia, it is not curative. Thus, additional treatment modalities are actively being investigated for this disease. In this context, chronic hypoxia treatment has shown some promise in various preclinical models of Friedreich’s ataxia, indicating a key role for oxygen in the pathogenesis173,174.
Fe-S transfer diseases include X-linked sideroblastic anemia with ataxia (XLSA/A), caused by a mutation in the ABCB7 transporter. The term ‘sideroblastic’ denotes the accumulation of iron-rich mitochondria ringing the nucleus in affected cells- ergo, mitochondria contain iron, but it is not utilized. XLSA/A is characterized by symptoms such as ataxia and cerebellar atrophy hypoplasia161,175,176, which usually present within the first 2 years of life. Six distinct mutations in ABCB7 have been reported to cause XLSA/A177,178,179,180. A deeper understanding of the substrate and transport mechanisms of ABCB7 is crucial to gain insight into the disease’s molecular underpinnings181,182.
Heme depletion
Heme, essential for oxygen transport, cellular respiration, and gene regulation, is mainly produced by erythroblasts and hepatocytes. Heme depletion can either be caused by environmental or genetic disruptions. Iron deficiency-induced anemia is estimated to affect one-third of the global population183. In the absence of iron, and subsequent depletion of heme, the HRI kinase is activated in red blood cells184,185. HRI acts to block global translation, while activating the ISR. Conversely, mutations in each of the eight heme biosynthetic enzymes cause different systemic implications30,186. The most common disorder is X-linked sideroblastic anemia (XLSA), caused by mutations in 5-aminolevulinic acid synthetase 2 (ALAS2), the initial rate-limiting step in erythroid heme synthesis. XLSA occurs mostly in males, with some cases in heterozygous female carriers. While pyridoxine, a cofactor for ALAS2, is a common treatment, not all patients respond to it. Recent studies have nominated DNA demethylating agents as a novel therapeutic approach187. Other enzyme defects in the heme biosynthesis pathway are associated with porphyria, classified as either erythropoietic or hepatic based on the site of intermediate product accumulation188.
Neurodegenerative diseases
Pathological iron accumulation is associated with several neurodegenerative diseases, including Alzheimer’s and Parkinson’s diseases. While these elevated iron levels are likely not the primary cause of these disorders, they may contribute to neuronal death. Moreover, the contributions of mitochondrial iron imbalances to neurodegeneration in these different disease contexts remains largely unexplored.
Alzheimer’s disease
Alzheimer’s disease (AD) is the most common form of dementia worldwide, estimated to account for 60–80% of cases189. AD is characterized by neuron degeneration in various cortical regions, leading to progressive memory loss and dementia. Indeed, changes in brain iron levels can already be detected in the early preclinical stages of AD190. In line with this finding, different measures of brain iron load have been shown to be prognostic biomarkers for predicted future cognitive decline and brain atrophy in AD191,192,193. Increased iron load in AD patients has been proposed to contribute to oxidative stress, both within neurons and glia, as well as amyloid plaque aggregation. Another AD marker, TAU, aggregates through heme oxygenase 1 overexpression and iron accumulation194. Dysregulated energy metabolism in the brain, driven by insulin resistance, is another key factor in AD progression and is linked to iron accumulation. This metabolic disruption contributes to the formation of amyloid plaques and tau pathology195,196. Given the role of iron accumulation in AD, iron chelators have been proposed as a potential therapeutic approach, with evidence suggesting they may inhibit amyloid aggregation197,198. Despite these indications, a randomized clinical trial investigating the effects of iron chelation on AD progression found that reducing iron levels accelerated cognitive decline and increased regional brain atrophy199. This surprising result raises the possibility that iron bioavailability is, in fact, limiting in AD.
Parkinson’s disease
Parkinson’s disease (PD), the most common neurodegenerative movement disorder, is characterized by accelerated neuronal death of primarily dopaminergic neurons and the accumulation of α-synuclein200. Already in 1923, German pathologist Friedrich Lewy described perivascular iron deposits in postmortem PD brains201. These findings have since been repeated many times over202,203,204,205. It has been proposed that PD iron overload can act as a driver of oxidative stress and iron-mediated cell death, i.e. ‘ferroptosis’. Moreover, iron and α-synuclein200 are connected both at the translational and protein level. First, the α-synuclein transcript was reported to contain an iron response element at its 5’UTR206, elevating its translation upon iron excess. Second, ferric iron can trigger α-synuclein aggregation207, and α-synuclein aggregates analyzed from postmortem brain samples are iron-laden208. However, in 2022, a PD clinical trial with the iron chelator deferiprone showed that, despite effectively reducing brain iron levels, deferiprone was associated with worsening motor and nonmotor symptoms209. In sum, while a link between iron accumulation and PD exists, the pathophysiological mechanisms of iron deposition and its effects on disease progression require additional study.
Mitochondrial iron and ferroptosis
Ferroptosis is an iron-dependent form of cell death, in which iron reacts with oxygen to drive the accumulation of lipid peroxides160. The oxidized plasma membrane becomes permeable to cations, culminating in cell swelling and plasma membrane rupture210,211,212. Multiple pathways can counteract the toxic accumulation of lipid peroxides, chief among them being the enzyme glutathione peroxidase 4 (GPX4). GPX4 can convert lipid hydroperoxides to less dangerous lipid alcohols, a reaction that depends on GSH213,214. Additional ferroptosis buffering pathways include ferroptosis suppressor protein 1 (FSP1), which regenerates reducing elements in the plasma membrane, such as CoQ or vitamin K, that can terminate the lipid peroxidation process. However, these ferroptotic-defense mechanisms can be overwhelmed, likely contributing to both rare and common pathologies, such as sedaghatian-type spondylometaphyseal dysplasia215, age-related macular degeneration216, and hereditary hemochromatosis217.
Given their central role in cellular iron and redox metabolism, it is not surprising that mitochondria are potent modulators of ferroptosis218. Mitochondria mediate CoQ biosynthesis and the breakdown of polyunsaturated fatty acids, which are the main source of possible lipid peroxides. CISD1, a 2Fe-2S cluster protein located in the mitochondrial outer membrane, protects against mitochondrial lipid peroxidation and thereby inhibits ferroptosis219. However, the mitochondria can also potentiate ferroptosis under some conditions. Upon cysteine deprivation, mitochondrial metabolism contributes to rapid glutathione depletion, leading to cellular lipid peroxide accumulation and subsequent ferroptosis220. In addition, disruptions in mitochondrial Fe-S cluster biosynthesis lead to the activation of the iron starvation response, elevating intracellular iron levels and the susceptibility to ferroptosis221,222. Taken together, the contribution of mitochondrial iron handling to ferroptosis is complex and likely context-dependent.
Future outlook
The past 25 years of research have revolutionized our understanding of the ways in which mitochondria metabolize iron, and how this metal, in turn, unlocks manifold abilities for the mitochondria. Future research will have to tackle important questions that remain unsolved: What is the entire spectrum of mitochondrial iron metalloproteins, whether they bind to Fe-S clusters, heme, or iron ions directly? These proteins are still being identified on an ad-hoc basis, and so we lack a comprehensive view of how iron sustains and regulates this ancient organelle. How is iron partitioned between the various pathways that it can support in the mitochondria? Does mitochondrial iron depletion induce additional signaling or restorative pathways, either within the mitochondria or extending into the cytosol? Can mitochondria communicate iron status directly to other organelles, e.g., the lysosome which functions as a key iron entry point? Can iron be exported from mitochondria? Other than in the context of heme, iron export pathways remain ill-defined. Why do certain tissues exhibit heightened vulnerability to mitochondrial iron dyshomeostasis? This variability likely stems from tissue-specific differences in iron demand, metabolic activity, regulation, and susceptibility to iron-induced cellular damage that requires further clarification. Deepening this understanding will provide an important framework for the development of therapeutic modalities that counteract iron-linked diseases.
Responses