Sex-specific regulation of the cardiac transcriptome by the protein phosphatase 2A regulatory subunit B55α

Introduction
Protein phosphatase 2A (PP2A) comprises a family of serine/threonine phosphatases with important roles in cardiac physiology and pathophysiology1. PP2A exists in two forms within the cell: as a heterodimer (the ‘core enzyme’) or as heterotrimers (active holoenzymes). The core enzyme consists of a catalytic ‘C’ subunit and a scaffolding ‘A’ subunit, each encoded by two isoforms (Cα/β and Aα/β). Association of the core enzyme with regulatory ‘B’ subunit isoforms gives rise to distinct PP2A holoenzymes, with different subcellular localisation patterns, substrate specificities and physiological functions2. More than 20 B subunit isoforms and splice variants have been identified, which can be grouped into four subfamilies: B/B55/PR55, B’/B56/PR61, B”/PR72 and B”’/striatins. Detailed study of PP2A B subunits in the heart is largely limited to a single isoform: B56α of the B’/B56 subfamily. In vitro and in vivo investigations have revealed information about its localisation in cardiomyocytes, key adaptor protein interactions and its role in cardiac contractility3,4,5,6,7,8. Investigation of other B subunit isoforms is critical for unravelling the complexity of PP2A signalling in the heart.
B55α of the B/B55 subfamily is a ubiquitously expressed PP2A regulatory subunit, with a high level of conservation between species at both the gene and protein level9,10,11. Its function in the heart has not been specifically investigated. We previously showed that B55α targets components of the PP2A core enzyme to histone deacetylase 5 (HDAC5) in adult rat ventricular myocytes12. This association increased with acute β-adrenergic receptor stimulation, leading to HDAC5 dephosphorylation, nuclear accumulation and repression of myocyte enhancer factor-2 (MEF2) transcriptional activity12. MEF2 transcription factors are essential for cardiogenesis and are critical regulators of cardiac hypertrophy and fibrosis in settings of cardiac stress/injury13,14,15,16. Studies in non-cardiac cell lines and tissues have reported that B55α dephosphorylates several other proteins with known roles in cardiac development and hypertrophy. These include Akt and FoxO117,18, key regulators of exercise-induced physiological cardiac hypertrophy19,20,21, and β-catenin22, a transcription factor involved in second heart field formation during embryogenesis and pathological cardiac hypertrophy induced by angiotensin II infusion or pressure overload23,24,25. Therefore, we hypothesised that B55α regulates cardiac development and hypertrophy in vivo.
As a first step to investigate B55α in the heart, we generated mice with global or cardiomyocyte-specific deletion of the gene encoding B55α (Ppp2r2a) and characterised the impact of reduced B55α expression on cardiac morphology, function and gene expression under basal conditions (i.e. in the absence of a physiological/pathological stress stimulus). Our findings validate previous work showing that B55α is critical for embryogenesis11,26 and provide the first characterisation of a B/B55 family member in the adult mouse heart. We demonstrate that cardiomyocyte B55α is not required for cardiac growth during embryonic development or the postnatal period. Reduced B55α expression did, however, lead to thinning of the left ventricular walls with ageing in male mice heterozygous for B55α (global knockout model). We also report a sex-specific role for B55α, with loss of cardiomyocyte B55α having a significant impact on the cardiac transcriptome in male mice, but not in female mice. These studies lay the foundation for future work investigating the role of B55α in cardiometabolic disease settings.
Methods
Experimental animals
Model 1: C57BL/6 N mice heterozygous (Het) for a Ppp2r2atm2a(KOMP)Wtsi ‘knockout-first’ allele (RRID: MMRRC:060623-UCD; see Fig. 1a) were generated by the Australian Phenomics Network (Monash University, Melbourne, Australia). Model 2: To generate cardiomyocyte-specific Ppp2r2a knockout (cKO) mice, mice heterozygous for the knockout-first allele were crossed with hemizygous transgenic mice expressing Flp recombinase (FlpTg/0 mice; RRID: IMSR_JAX:003800; see Fig. 1a). This removed the lacZ reporter and neomycin selection cassette and converted Ppp2r2atm2a(KOMP)Wtsi to a floxed Ppp2r2a allele. Next, offspring hemizygous for Flp and heterozygous for the floxed Ppp2r2a allele (FlpTg/0 Ppp2r2aL/+) were crossed to remove the Flp transgene. Flp-negative floxed Ppp2r2a mice (Flp0/0 Ppp2r2aL/L) were then mated with hemizygous transgenic mice expressing Cre recombinase under the control of the αMHC promotor (αMHC-CreTg/0 mice, FVB background)20,27. Importantly, Cre expression in this strain does not lead to cardiotoxicity20,28. CreTg/0 Ppp2r2aL/+ and Cre0/0 Ppp2r2aL/+ mice were mated to produce the following 6 cardiomyocyte-specific genotypes: CreTg/0 Ppp2r2aL/L (B55α knockout, cKO), Cre0/0 Ppp2r2aL/L (floxed control, FC), CreTg/0 Ppp2r2aL/+ (B55α heterozygote, cHET), Cre0/0 Ppp2r2aL/+ (half-floxed control), CreTg/0 Ppp2r2a+/+ (Cre control), Cre0/0 Ppp2r2a+/+ (wildtype control). FC and cKO mice for the RNA-seq analysis were generated from CreTg/0 Ppp2r2aL/L x Cre0/0 Ppp2r2aL/L breeding pairs. All aspects of animal care and experimentation were approved by The Alfred Research Alliance Animal Ethics Committee. Mice were housed in a temperature-controlled environment with a 12-hour light/dark cycle and fed standard chow ad libitum.

a Schematic of the Ppp2r2a ‘knockout-first’ allele used to produce B55α heterozygous (Het) and homozygous (Hom) knockout mice (Model 1: Global B55a KO). Floxed Ppp2r2a mice were produced by removing the lacZ/neo cassette with Flp recombinase. Cardiomyocyte-specific B55α knockout mice (Model 2) were produced by removing exon 4 with Cre recombinase under control of the αMHC promoter. b Whole mount images of wildtype (Wt) and Hom littermates at 12.5 dpc, imaged using an AxioCamMRc5 camera with a 1x objective. Scale bar 2 mm. c Images of Wt and Hom embryos at 12.5 dpc, stained with haematoxylin & eosin and imaged using a Pannoramic Scan II slide scanner fitted with a 20x/NA 0.8 objective and a CCD camera. Left panel shows head (scale bar 1 mm), middle panel shows hindlimb (scale bar 0.5 mm), right panel shows skin layer (scale bar 0.05 mm). d Images of Wt and Hom hearts at 12.5 dpc, stained with haematoxylin & eosin and imaged using a Pannoramic Scan II slide scanner fitted with a 20x/NA 0.8 objective and a CCD camera. Left panel shows heart cross-section (scale bar 0.5 mm; asterisks indicate AV cushions, v = ventricle), right panel shows ventricular wall (scale bar 0.05 mm; c = compact myocardium, t = trabeculae).
Timed matings for collection of embryos at 12.5 dpc
Het breeding pairs were mated. Pregnancy was identified via the presence of a vaginal plug, and midday was designated as 0.5 days post coitum (dpc). Pregnant females were anesthetised with pentobarbital (~30 mg intraperitoneal) and euthanised via cervical dislocation. An incision was made in the lower abdomen, and the uterine horn removed and immediately placed on sterile gauze in an ice slurry. Embryos were carefully dissected under microscope guidance and placed in cold 4% paraformaldehyde (PFA) with gentle shaking overnight.
Embryo histology
Embryos were dehydrated, paraffin-embedded in a sagittal orientation and 5 µm sections obtained at the level of the heart. Sections were stained with hematoxylin & eosin (H&E) and imaged using a Pannoramic Scan II (3D Histech, Budapest, Hungary), using a Carl Zeiss Plan-Apochromat 20x/NA 0.8 objective and a Point Grey Grasshopper 3 CCD monochrome camera. Digital slide images were viewed using SlideViewer software version 2.6 (3D Histech, Budapest, Hungary). Embedding and processing was conducted by the University of Melbourne Histology Platform (Melbourne, Australia) and slide scanning performed by Phenomics Australia (Melbourne, Australia). Additionally, four paraffin-embedded embryos (3 Hom, 1 Wt) were submitted to Phenomics Australia for histopathological evaluation29,30,31,32,33,34 by two independent pathologists.
Echocardiography
Echocardiography was performed on anaesthetised mice (1.8% isoflurane) using either an iE33 ultrasound machine with a 15-megahertz (mHz) linear array transducer (Phillips, Amsterdam, Netherlands) or a Vevo 2100 High-Frequency Ultrasound System with a MS550D transducer (Visual Sonics, Toronto, ON, Canada). Short axis M-mode images were used to measure interventricular septum (IVS) and left ventricular posterior wall (LVPW) thicknesses at diastole, as well as internal dimensions at diastole (LVID; d) and systole (LVID; s). Dimensions were measured from three beats per echocardiogram and averaged. Fractional shortening and LV mass were calculated using the following equations, respectively: [(LVID; d – LVID; s)/LVID; d] x 100%, [(IVS + LVPW + LVID; d)3 – LVID; d 3]. Investigators were blinded to genotype during image acquisition and analysis. All analyses were independently validated by the Baker Heart & Diabetes Institute Preclinical Microsurgery & Imaging Platform (Melbourne, Australia)35.
Tissue collection
Adult mice were anesthetised with pentobarbital (~30 mg intraperitoneal) and euthanised via cervical dislocation. The heart was quickly removed, washed in cold PBS, excess moisture removed with sterile gauze, and weighed. The atria were dissected and weighed. Ventricles were cut along the short axis, separating the base and apex. The base was fixed in cold 4% PFA for processing and paraffin embedding. The apex was divided evenly along the long axis and snap-frozen in liquid nitrogen for protein and RNA extraction. Other tissues including lungs, liver, kidney, and spleen were weighed and snap-frozen in liquid nitrogen.
Gene expression by quantitative PCR
Total RNA was extracted from ventricles using TRI-Reagent (Sigma Aldrich, St. Louis, MO, US), according to the manufacturer’s instructions. Nanodrop spectrophotometry (Thermo Scientific, Waltham, MA) was used to quantify RNA. A High-Capacity RNA-to-cDNA kit (Thermo Scientific, Waltham, MA) was used to reverse transcribe 2 µg RNA to cDNA according to the manufacturer’s instructions. RT-qPCR was conducted using Taqman gene expression assays and Taqman Fast Universal Master Mix (Thermo Fisher Scientific, Waltham, MA) or primers and SYBR Green PCR Master Mix (Thermo Fisher Scientific, Waltham, MA). Targets were amplified using a Quant Studio ViiA7 Real-Time PCR System. The 2–ΔΔCt method was used for the calculation of fold difference using hypoxanthine phosphoribosyltransferase 1 (Hprt1) as a reference gene. For SYBR Green reactions, primer efficiencies were calculated according to the following equation: E = 10[-1/slope], using the slope of a standard curve generated using serial dilutions of cDNA. Melt curves were performed to assess the specificity of the amplification product. Samples were run in triplicate, and samples with replicates with a standard deviation exceeding 0.5 were excluded. Taqman gene expression assays from Thermo Fisher Scientific (Waltham, MA) were as follows: Nppa Mm01255747_g1, Nppb Mm01255770_g1, Myh6 Mm00440359, Myh7 Mm01319006, Col1a1 Mm00801666_g1, Col3a1 Mm00802300_m1, Ctgf Mm01192932_g1, Hprt1 Mm01545399_m1. SYBR Green primer sequences were as follows: Ppp2r2a fwd 5’-CCGTGGAGACATACCAGGTA-3’, rev 5’-AACACTGTCAGACCCATTCC-3’; Ppp2r2c fwd 5’-AGCGGGAACCAGAGAGTAAG-3’, rev 5’-GTAGTCAAACTCCGGCTCG-3’; Ppp2r2d fwd 5’- TTACGGCACTACGGGTTCCA-3’, rev 5’-TTCGTCGTGGACTTGCTTCT-3’; Hprt1 fwd 5’- TCCTCCTCAGACCGCTTTT-3’, rev 5’-CCTGGTTCATCATCGCTAATC’-3’.
Protein analysis
Ventricles were homogenised in lysis buffer [20 mM Tris-HCl (pH 7.4), 127 mM NaCl, 10% glycerol, 1% IGEPAL, 20 mM NaF, 10 mM EGTA, 1 mM sodium pyrophosphate, 1 mM vanadate, 1 mM PMSF, 4 µg/mL pepstatin, 4 µg/mL aprotinin, 4 µg/mL leupeptin], incubated on ice for 15 min, and centrifuged at 4 °C for 15 min at 16,000 g to pellet cellular debris. Protein lysates (80 µg) and molecular weight marker (Precision Plus Protein All Blue Prestained Protein Standard, Bio-Rad, 1610373) were separated using 7.5% or 10% SDS-PAGE gels, transferred to PVDF membranes, and incubated with primary antibody overnight at 4°C or 1.5 hours at room temperature for HRP-conjugated secondary antibodies. Chemiluminescent signals were quantified using ImageJ pixel analysis (US National Institutes of Health) or Genetools analysis software (Syngene, Cambridge, UK). Data were expressed as fold difference relative to the control group. Antibodies were as follows: B55α (#sc-81606, Santa Cruz, 1:1000), FoxO1 (#2880, Cell Signaling Technology, 1:2000), pSer256 FoxO1 (#9461, Cell Signaling Technology, 1:1000), pThr24/Thr32 FoxO1/O3a (#9464, Cell Signaling Technology, 1:2000), HDAC5 (#20458, Cell Signaling Technology, 1:500), pSer259 HDAC5 (#3443, Cell Signaling Technology, 1:500), PP2A-C (#2038, Cell Signaling Technology, 1:1000), PP2A-A (#2039, Cell Signaling Technology, 1:250), α-tubulin (#2144, Cell Signaling Technology, 1:2500) and vinculin (#V9131, Sigma Aldrich, 1:5000). All uncropped blots are included in the Data Supplement.
RNA-sequencing
Left ventricle tissue (~20 mg) was mechanically homogenised in TRI-Reagent with a 5 mm steel bead at 30 Hz for 2 × 30 s (TissueLyser II, Qiagen) then centrifuged for 10 min at 10,000 g. RNA was extracted from the supernatant using spin columns with DNase-I treatment (Zymo DirectZol RNA Miniprep kit #R2050, Integrated Sciences, Australia). RNA concentration and purity was determined spectrophotometrically (Nanodrop, Thermo Fisher, Australia) and RNA integrity was determined by electrophoresis (Tapestation, Agilent). RNA integrity (RIN) values for all samples were ≥7.7. Stranded libraries were prepared from 500 ng total RNA input with poly-A+ selection (TruSeq Stranded mRNA, Illumina). Library sizes and concentrations were confirmed prior to sequencing (TapeStation, Agilent). At least 40 million 150 bp paired-end reads per library were generated on the Illumina NovaSeq 6000 platform (Australian Genome Research Facility, Melbourne, Australia). Raw read preprocessing was conducted on the Galaxy Australia platform36. Reads underwent initial quality check with FastQC v0.74, then quality filtering and adapter trimming was conducted with Cutadapt v4.6. Filtered reads were aligned to the mouse reference genome (Mus musculus, Ensembl version GRCm39.104) using STAR v2.7.11a37 in 2-pass mode. Gene expression was quantified at the gene level using HTseq-counts v2.0.5 and collated into a counts matrix. Analysis of differential expression was performed on genes with ≥1 counts per million (CPM) in all samples in DESeq2 in iDEP v2.038. False discovery rate (FDR) was calculated using Benjamini-Hochberg method and genes with an FDR < 0.05 were considered differentially expressed (DE). DE genes were analysed for pathway enrichment using Gene Ontology (GO) genesets in iDEP v2.0.
Fibrosis analysis
Paraffin-embedded left ventricles were cut (6 μm sections), stained with picrosirius red and imaged using a Pannoramic Scan II (3D Histech, Hungary) using a Carl Zeiss Plan-Apochromat 20x/NA 0.8 objective and a Point Grey Grasshopper 3 CCD monochrome camera. Images of scanned sections were obtained at 20x magnification (10 regions/heart) using SlideViewer software version 2.6 (3D Histech, Budapest, Hungary). The number of red pixels (collagen) was counted using ImageJ (NIH, Bethesda, USA) and divided by tissue area to calculate the percentage of fibrosis. Analysis was undertaken blinded to sex and genotype.
Data presentation and statistical analysis
Data are presented as scatterplots with bars and lines indicating the mean +/− SEM (for qPCR data, as per Thermo Fisher Scientific’s Guide to Performing Relative Quantitation of Gene Expression Using Real-Time Quantitative PCR, 2008 edition) or median +/− IQR (all other data). Fold difference (qPCR and Western blot data) was calculated by dividing by the mean or median of the control group, as appropriate. Gene expression data were analysed using two-sided unpaired t-tests (2 groups) or two-way ANOVA followed by Tukey’s or Sidak’s post hoc tests, as appropriate. All other data were analysed using Mann-Whitney U tests (2 groups) or Kruskal-Wallis tests (6 groups). A P-value < 0.05 was deemed statistically significant. Animal numbers and data exclusions are reported in Supp Fig. 1.
Results
To determine if PP2A-B55α regulates heart size or morphology in vivo, we first generated mice with heterozygous (Het) or homozygous (Hom) global disruption of Ppp2r2a (Model 1, Fig. 1a). The knockout-first allele has conditional potential due to the presence of FRT and loxP sites, allowing removal of the lacZ reporter and neomycin selection cassette to generate floxed Ppp2r2a alleles for cardiomyocyte-specific Cre-mediated deletion of exon 4 (Model 2, Fig. 1a).
Homozygous knockout of B55α causes embryonic lethality without overt cardiac defects
The expected Mendelian ratio of Wt : Het : Hom mice in the global knockout mouse strain (Model 1) was 1:2:1. However, no Hom offspring were born to Het breeding pairs, indicating lethality and resorption of Hom embryos in utero. Consistent with previous reports in different B55α knockout mouse strains11,26, Hom embryos were smaller than Wt littermates at 12.5 dpc (Fig. 1b) and started dying between 12.5 and 14.5 dpc (26% Hom at 12.5 dpc, 6% Hom at 14.5 dpc). Histopathological evaluation at 12.5 dpc identified several abnormalities in Hom embryos, including arrested oral/nasal cavity development (Fig. 1c, left panel), arrested limb bud development (Fig. 1c, middle panel), and deficient skin layers (Fig. 1c, right panel). One of the three Hom embryos evaluated also had a smaller liver and displayed hepatocyte necrosis. Hearts of Hom embryos were assessed for the presence of congenital cardiac abnormalities, including atrial or ventricular septal defects, abnormal leaflet formation or myocardial non-compaction, however there were no obvious micromorphological differences compared with Wt (Fig. 1d).
Reduced expression of B55α did not lead to left ventricular hypertrophy or dysfunction in young mice but induced LV wall thinning in male mice with age
Ventricular B55α protein levels were ~45% lower in the male Het group vs Wt and ~70% lower in the female Het group vs Wt (Fig. 2a, b), allowing us to investigate the impact of reduced B55α expression on cardiac morphology and function in adult mice. There were no differences in the relative abundance of B55α between male and female control hearts (Supp Fig. 2). We assessed mice at 10–12 weeks of age (representing early adulthood) and at 12 months (representing middle age). By echocardiography, there were no differences in left ventricular wall thicknesses or internal dimensions between Wt and Het mice in the 10–12-week old cohort (Supp Table 1). Heart rate and fractional shortening, a measure of left ventricular contractility, were also not different between Wt and Het mice (Supp Table 1). At 12 months of age, male Het mice had thinner LV walls compared to Wt mice based on LVPW and IVS thicknesses (Fig. 2c). This phenotype was less apparent in females, with lower IVS thickness in Het mice but no difference in LVPW (Fig. 2d). No other differences in echocardiographic parameters were observed (Fig. 2c, d; Supp Table 2).
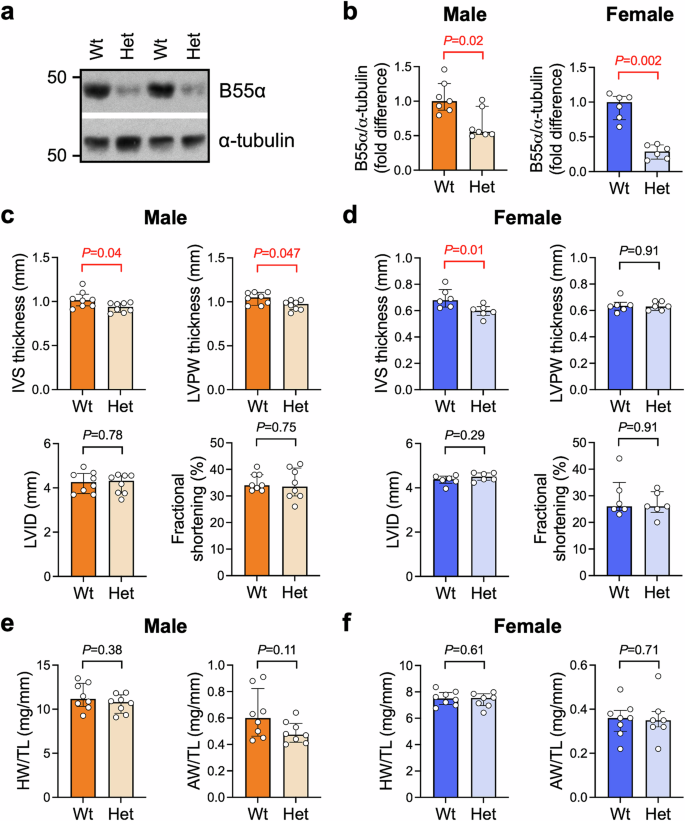
a Representative Western blot and b) quantitation of B55α protein expression. Ventricular tissue from 12-month old male and female Ppp2r2a wildtype (Wt) and heterozygous (Het) mice at 12 months of age. α-tubulin was used as a loading control. Data are expressed as fold difference vs Wt. Bars show median +/− IQR. Mann-Whitney U tests. n = 6–7 per group. c, d Interventricular septum (IVS) thickness, left ventricular posterior wall (LVPW) thickness, LV internal dimension (LVID) and fractional shortening, assessed by M-mode echocardiography in anaesthetised mice at 12 months of age. Bars show median +/− IQR. Mann-Whitney U tests. n = 6–8/group. e, f Heart weight (HW) and atria weight (AW) normalised to tibia length (TL) of mice at 12 months of age. n = 6-8/group. Bars show median +/− IQR. Mann-Whitney U tests.
Assessment of organ weights at 10–12 weeks and at 12 months of age revealed no differences in heart weight or atria when normalised to body weight or tibia length, although atria tended to be smaller in male Het vs Wt in the younger cohort (Fig. 2e, f; Supp Table 3, Supp Table 4). Lung weight was within the normal range, consistent with the absence of cardiac pathology (which can lead to pulmonary congestion; Supp Tables 3, 4). Liver weight was lower in female Het vs Wt in the younger cohort (Supp Table 3) and tended to be higher in male Het vs Wt in the older cohort (see Supp Table 4). Variations in the liver were also observed in Hom embryos at 12.5 dpc (section “Homozygous knockout of B55α causes embryonic lethality without overt cardiac defects”), indicating a possible hepatic function for B55α. We did not measure blood pressure in these mice, which is a limitation of the study.
To investigate if the mild LV wall thinning observed in Het mice at 12 months of age was accompanied by differences in the expression of genes involved in pathological cardiac remodelling, we performed qPCR on ventricular lysates (Supp Fig. 3a, b). Levels of cardiac stress markers atrial and B-type natriuretic peptides (ANP and BNP, encoded by Nppa and Nppb, respectively) did not differ in Het mice compared to Wt in either sex. There were no differences in the expression of myosin heavy chain isoforms αMHC or βMHC (encoded by Myh6 and Myh7, respectively). Expression of type I and III collagens (Col1a1 and Col3a1) and connective tissue growth factor (Ctgf) were comparable between genotypes, suggesting an absence of cardiac fibrosis. We also assessed the expression of blood and lymphatic vessel markers (Cd31 and Lyve1, respectively) as impaired angiogenesis or lymphangiogenesis in the heart is deleterious39,40, and a previous study reported vascular and lymphatic vessel defects in skin of embryos lacking B55α11. We observed a significant reduction in mRNA, but not protein, levels of the lymphatic vessel marker Lyve1 in male Het ventricles vs Wt (Supp Fig. 3c, d), while Cd31 expression was not different between genotypes (Supp Fig. 3c). Lyve1 mRNA and protein levels were not different in female Het and Wt hearts (Supp Fig. 3e, f). Cd31 mRNA expression tended to be lower in female Het mice vs Wt, however the effect was small (~10% decrease, P = 0.05, Supp Fig. 3e).
Loss of B55α in cardiac myocytes does not impact postnatal cardiac growth, but alters the transcriptional profile in male mice
To circumvent the effects of B55α deletion on embryonic development and investigate the role of cardiomyocyte B55α in postnatal heart growth, we next characterised the phenotype of mice with cardiomyocyte-specific knockout (cKO) of B55α using an αMHC-Cre line (Model 2, Fig. 1a). We confirmed loss of B55α in ventricular tissue of 10–12-week old cKO mice by Western blotting. As expected, B55α protein levels were significantly lower (~90% decrease in expression) in both male and female cKO hearts compared to floxed controls (FC, Fig. 3a–d). We also examined the expression of the other components of the heterotrimeric PP2A holoenzyme. We observed a significant decrease (~35%, P = 0.03) in PP2A-A protein levels in female cKO hearts, but no differences in the expression of PP2A-C (Fig. 3a–d). Deletion of Ppp2r2a in the heart was not associated with differences in the expression of other PP2A B55 subunit isoforms, i.e. Ppp2r2c (encoding B55γ) or Ppp2r2d (encoding B55δ; Fig. 3e, f). We also observed no differences in HDAC5 phosphorylation at Ser259 between genotypes in either sex (Fig. 3g, h), suggesting that HDAC5 is not phospho-regulated by PP2A-B55α in the heart under normal physiological conditions.
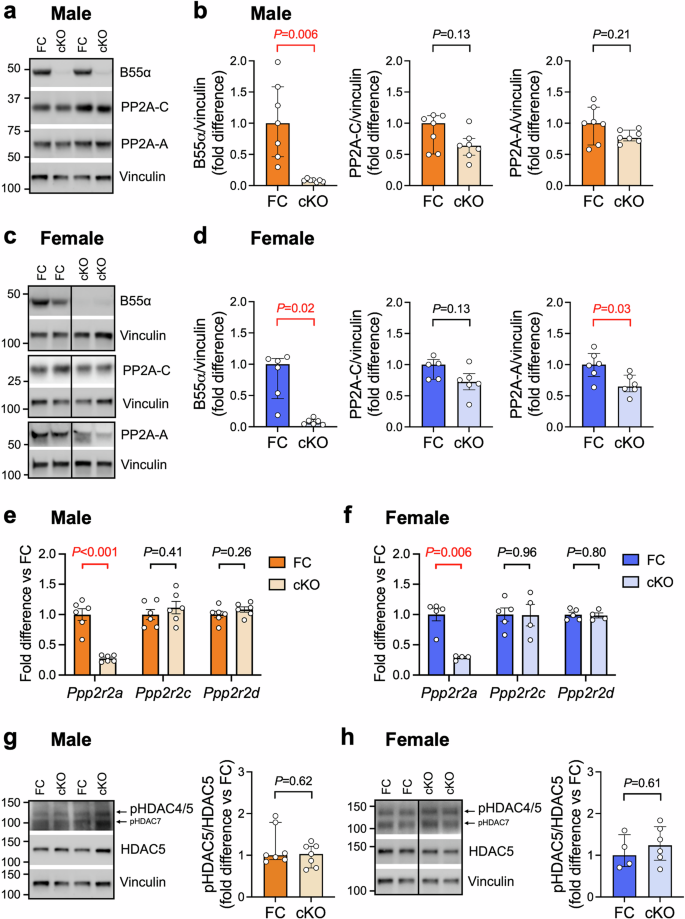
a, c Representative Western blots and b, d quantitation of B55α, PP2A catalytic (PP2A-C) and scaffolding (PP2A-A) subunits in ventricular tissue from 10–12 week-old cardiomyocyte-specific B55α knockout (cKO) mice and floxed controls (FC). Vinculin was used as a loading control. Data are expressed as fold difference vs FC. Bars show median +/− IQR. Mann-Whitney U tests. n = 6 per group for all analyses except PP2A-C (n = 5 for female FC). Vertical lines indicate where images of Western blots have been spliced so the relevant genotypes could be displayed side by side. e, f Quantitative PCR analysis of PP2A B55 subunits Ppp2r2a (encoding B55α), Ppp2r2c (encoding B55γ) and Ppp2r2d (encoding B55δ). Data are expressed as fold difference relative to the FC group. Bars show mean +/− SEM. Unpaired t-tests. n = 4–6/group. g, h Representative Western blots and quantitation of HDAC5 phosphorylation at Ser259 relative to total HDAC5. Data are expressed as fold difference vs FC. Bars show median +/− IQR. Mann-Whitney U tests. n = 4–7 per group.
At 10–12 weeks of age, there were no detectable differences in LV wall thicknesses or chamber dimensions between any of the genotypes (Fig. 4a, b; Supp Table 5). Fractional shortening tended to be lower in male cKO vs FC, but the effect was small (~10% decrease, P = 0.10). Heart, atria and lung weight of both male and female cKO mice were comparable with controls (Fig. 4c, d; Supp Table 6), indicating the absence of pathological cardiac hypertrophy and dysfunction.
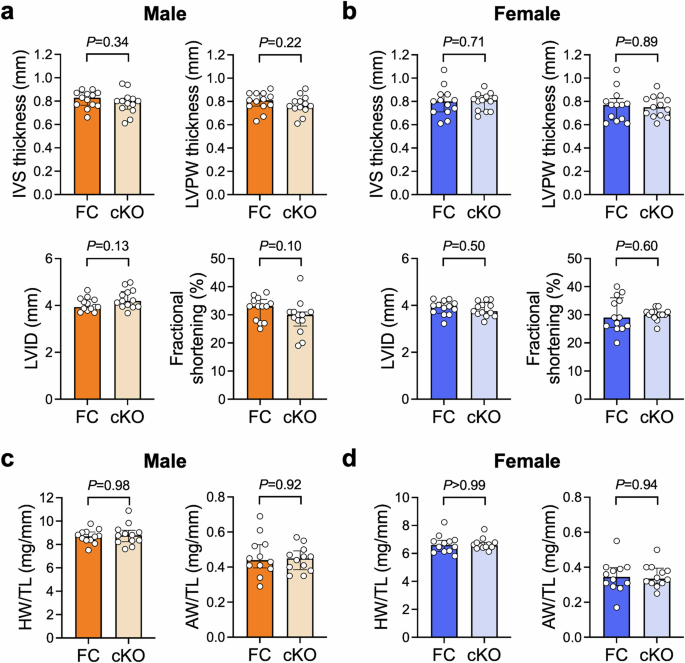
a, b Interventricular septum (IVS) thickness, left ventricular posterior wall (LVPW) thickness, LV internal dimension (LVID) and fractional shortening in 10–12 week old cardiomyocyte-specific B55α knockout (cKO) mice and floxed controls (FC), quantified from M-mode echocardiograms. Bars show median +/- IQR. Mann-Whitney U tests. n = 13/group. c, d Heart weight (HW) and atria weight (AW) normalised to tibia length (TL) in FC and cKO mice at 10–12 weeks of age. Bars show median +/- IQR. Mann-Whitney U tests. n = 12/group.
Interestingly, we observed differences in the expression of genes involved in extracellular matrix and myofilament composition in ventricular tissue of male mice (Fig. 5a). Expression of type I collagen (Col1a1) and connective tissue growth factor (Ctgf) were modestly increased in male cKO hearts compared with FC (~30% increase in Col1a1, P = 0.04; ~70% increase in Ctgf, P = 0.02, Fig. 5a). Type III collagen expression also tended to be higher (~20% increase in Col3a1, P = 0.08, Fig. 5a). Myh7 (encoding βMHC) was also significantly higher in male cKO hearts vs FC (~70% increase, P = 0.02, Fig. 5a). Upregulation of the foetal βMHC isoform is frequently observed in cardiac disease settings16,41,42. Coupled with our observation of lower fractional shortening in male cKO mice vs FC, this gene expression profile could indicate the early onset of pathology in male cKO mice, although increased expression of the βMHC isoform is typically accompanied by a reciprocal decrease in the expression of the αMHC isoform.
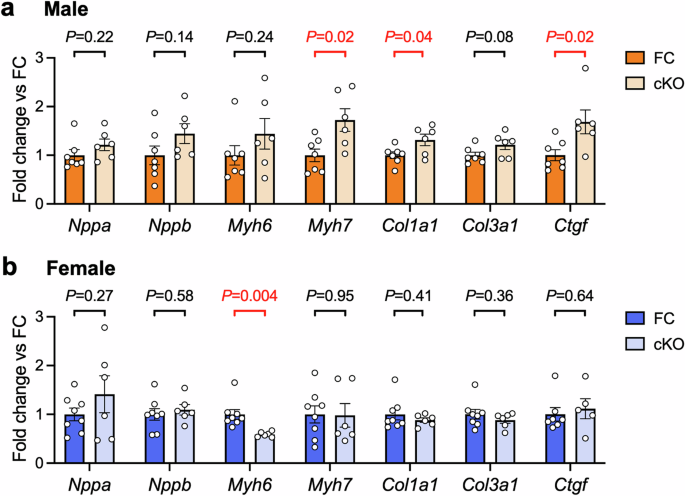
a, b Quantitative PCR analysis of genes that are typically dysregulated in settings of pathological cardiac hypertrophy and fibrosis. Ventricular tissue from male and female cardiomyocyte-specific B55α knockout (cKO) and floxed control (FC) mice. Data are expressed as fold difference relative to the FC group. Bars show mean +/− SEM. Unpaired t-tests. n = 6–8 per group for all analyses except Ctgf (n = 5 for female cKO).
We and others have previously shown that the αMHC-Cre transgenic mouse strain used in this study does not have increased collagen expression or fibrosis at 6-8 months of age20,28. However, as another αMHC-Cre strain (with much higher levels of Cre expression) develop cardiac fibrosis28, we compared Col1a1, Col3a1 and Ctgf expression in hearts of male FC, cKO and Cre control mice. Our analyses revealed no differences in expression between the FC and Cre control groups (Supp Fig. 4), confirming that the modest increases in expression observed in cKO hearts were due to deletion of B55α and not an off-target effect of Cre expression.
In female cKO mice, increases in fibrotic gene expression were not present (Fig. 5b). We observed reduced expression of Myh6, a known MEF2 transcriptional target encoding the dominant adult αMHC isoform (~40% decrease, P = 0.004, Fig. 5b). Expression of the natriuretic peptide genes Nppa and Nppb were not different in male or female cKO hearts compared to control.
To further investigate differences in cardiac gene expression arising from loss of cardiomyocyte B55α, we performed RNA-seq analysis on a separate cohort of FC and cKO mice. Interestingly, and consistent with the findings of our qPCR analysis (Fig. 5a, b), we observed a greater number of genes that were differentially expressed due to B55α deletion in the male heart compared with the female heart (Fig. 6a, b; Supp Table 7). Remarkably, only 5 genes (in addition to Ppp2r2a) were affected by B55α deletion in the female heart (Fig. 6b), while 543 were differentially expressed in male cKO vs FC (FDR < 0.05, Fig. 6a). Eya1 was the only gene that was significantly upregulated in both male and female hearts with B55α deletion (Fig. 6a, b). Eya family members are transcriptional co-activators that have been shown to physically interact with B55α, directing PP2A activity towards transcription factors such as c-Myc43. The physiological significance of this interaction has not been investigated in the heart.
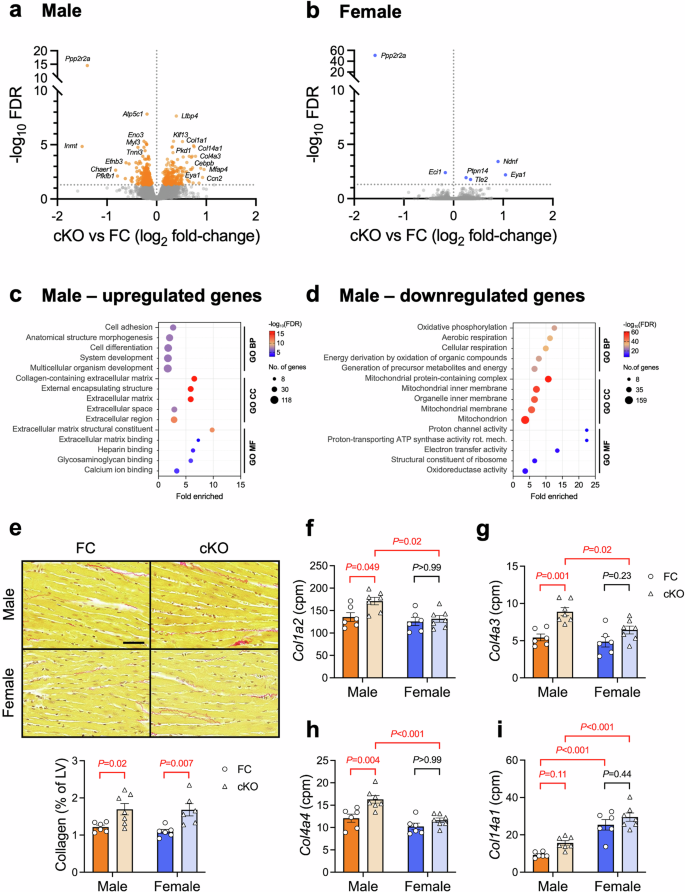
a, b Volcano plots showing genes that were differentially expressed between cKO and FC hearts. RNA-seq analysis of ventricular tissue from male and female cardiomyocyte-specific B55α knockout (cKO) and floxed control (FC) mice. FDR < 0.05, n = 6-7/group. c, d Pathway enrichment analysis showing Gene Ontology terms that were over-represented in the set of genes that were significantly upregulated or downregulated in male cKO hearts vs FC. n = 6–7/group. e Representative LV sections stained with picrosirius red and quantitation of collagen (expressed as a percentage of left ventricular area). Scale bar: 50 μm. Two-way ANOVA followed by Sidak’s post hoc tests. n = 6-7/group. f–i Relative mRNA abundance (in counts per million, cpm) of collagen isoforms in FC (circles) and cKO (triangles) hearts. Two-way ANOVA followed by Tukey’s posthoc tests. n = 6–7/group.
Pathway enrichment analysis of genes that were upregulated in male cKO vs FC hearts revealed an over-representation of Gene Ontology terms associated with cell/tissue/organism development and morphogenesis (Biological Process), extracellular matrix (Cellular Component), and ECM structural constituents and binding (Molecular Function, Fig. 6c; Supp Table 8). Amongst genes that were downregulated in male cKO vs FC hearts, there was an over-representation of terms associated with oxidative phosphorylation and energy production (Biological Process), mitochondria (Cellular Component), and activity of the electron transport chain (Molecular Function, Fig. 6d). These data suggest a role for B55α in the maintenance of ECM composition and mitochondrial energy metabolism in the male heart.
Analysis of LV cross-sections stained with picrosirius red revealed that upregulation of ECM-associated genes in male cKO mice translated to a small increase in collagen protein deposition (Fig. 6e). Surprisingly, we detected a similar increase in LV collagen content in female cKO mice vs FC, despite no differences in the expression of Col1a1 or Col3a1 (Figs. 5b and 6b). Examination of other collagen species quantified by RNA-seq revealed a significant effect of genotype and sex for Col1a2, Col4a3, Col4a4 and Col14a1 (P < 0.05 for all, Fig. 6f–i). Col4a3 and Col4a4 expression tended to be higher in female cKO hearts vs FC, although these differences were not as pronounced as in male hearts and were not statistically significant (Fig. 6g,h). Col14a1 expression was significantly higher in female hearts compared with male hearts (~2.7-fold increase in female FC vs male FC, P < 0.001, Fig. 6i). Collectively, these data identify cardiomyocyte B55α as a regulator of the cardiac ECM.
In addition, we investigated the phosphorylation status of FoxO1, a reported target of B55α (at Ser256 and Thr24)18 and key transcription factor regulating mitochondrial homeostasis and cardiac energy metabolism44,45. FoxO1-dependent gene transcription is regulated by phosphorylation, with Ser256 dephosphorylation promoting the nuclear accumulation of FoxO1 and Ser256 phosphorylation promoting cytosolic accumulation in cardiomyocytes46. We detected no differences in the phosphorylation of FoxO1 at Ser256 or Thr24 between genotypes (Supp Fig. 5), suggesting that FoxO1 is unlikely to have contributed to the gene expression differences we observed in hearts of B55α cKO mice.
Discussion
The aim of this study was to determine if the PP2A regulatory subunit B55α is required for heart growth during normal physiological development. We generated mice with global or cardiomyocyte-specific deletion of B55α and examined the cardiac phenotype during embryogenesis, early adulthood (10–12 weeks) and at 12 months of age. The key findings of this study are: 1) B55α is not required for physiological heart growth during embryogenesis or the postnatal period; 2) in male mice, reduced B55α expression leads to mild thinning of the ventricular walls with ageing; 3) cardiomyocyte-specific deletion of B55α in male mice increases the expression of genes involved in extracellular matrix composition and decreases the expression of genes involved in mitochondrial energy production; 4) loss of B55α increases collagen accumulation in both male and female mouse hearts; and, 5) under basal conditions, B55α is not a major regulator of the female cardiac transcriptome. Collectively, these data demonstrate that the heart can maintain a relatively normal growth trajectory and function, even if B55α protein levels are markedly reduced. This may have implications for PP2A-targeting therapeutics (discussed below). Our data reveal sex-specific roles for B55α in cardiomyocytes and point to a possible role for B55α signalling in cardiac endothelial cells during ageing.
Our finding of embryonic lethality in Hom embryos is consistent with two previous publications11,26. Mice with CRISPR/Cas9-mediated deletion of Ppp2r2a exon 4 die between 10.5 dpc and birth, with epidermal defects visible as early as 10.5 dpc and limb and digit defects evident at later stages26. Mice with PGK-Cre-mediated (i.e. ubiquitous) deletion of exon 6 die between 12.5 and 15.5 dpc, and display reduced blood and lymphatic vessel density in skin11. This was attributed to loss of B55α in endothelial cells, as conditional deletion of B55α in this cell type recapitulated the phenotype11. Neither study examined the cardiac phenotype of B55α knockout embryos.
In the current study, the cardiac phenotype of embryos with homozygous deletion of B55α was assessed at 12.5 dpc, a developmental timepoint when atrial and ventricular septation is occurring and the ventricular walls are thickening due to myocardial cell proliferation32. Evaluation of H&E-stained sections at 50 μm intervals spanning the heart revealed no overt signs of abnormal cardiac development in Hom embryos. It is possible that more detailed investigations could reveal subtle phenotypic changes that are not detectable by H&E staining. Based on the findings of Ehling and colleagues11, it would not be surprising if hearts of knockout mice displayed reduced blood and lymphatic vessel density. Endothelial cells are the most abundant non-myocyte cell type in the heart47, and the development of both the coronary and lymphatic vasculature commences prior to embryonic day 14.5, coinciding with the occurrence of embryonic lethality in our model48,49,50. In support of a role for B55α in cardiac endothelial cell populations, we observed a decrease in mRNA levels of the lymphatic vessel marker Lyve1 in hearts of male mice with reduced B55α expression at 12 months of age, although this was not associated with a detectable reduction in LYVE1 protein. In the heart, lymphatic vasculature is critical for the maintenance of fluid equilibrium as well as nutrient and immune cell transport39,40,49,50,51,52, and Lyve1 knockout mice had worse cardiac function and more pronounced wall thinning than control mice in response to myocardial infarction39. Thus, reduced B55α in endothelial cell populations may make the heart more susceptible to pathological cardiac remodelling and dysfunction with ageing or in settings of cardiac stress or injury.
After birth, the heart responds to sustained increases in workload by increasing heart mass53. This is achieved via the enlargement of heart muscle cells (cardiomyocyte hypertrophy). The signalling pathways regulating cardiomyocyte hypertrophy are complex, involving numerous receptors and stretch sensors, signalling mediators and transcription factors. A key signalling axis regulating cardiomyocyte hypertrophy in disease settings is the HDAC5/MEF2 axis54. The adult heart expresses MEF2A and MEF2D isoforms, which form homodimers or heterodimers with other transcription factors (e.g. GATA, NFAT and STAT3) and bind to DNA promoter regions to modulate gene transcription55,56,57. A seminal study from the Olson group showed that MEF2D activation is sufficient and necessary for pathological cardiac remodelling16, while MEF2A may play a protective or a pathological role58,59. The subcellular distribution of HDAC5 is a key determinant of MEF2 transcriptional activity. When localised to the nucleus, HDAC5 represses MEF2. Nuclear export and cytosolic accumulation of HDAC5 following phosphorylation by protein kinases alleviates the repressive interaction with MEF2 and facilitates the transcription of MEF2-dependent genes60,61,62,63,64,65.
We previously showed that B55α targets the PP2A core enzyme to HDAC5 in cardiomyocytes12. We hypothesised that loss of B55α would lead to cardiac hypertrophy, i.e. via hyper-phosphorylation of HDAC5 and activation of MEF2. However, we observed no effect of cardiomyocyte-specific deletion of B55α on LV morphology or heart weight in male or female mice at 10–12 weeks of age. We also observed no differences in the phosphorylation of HDAC5 at Ser259, a key determinant of HDAC5 subcellular localisation which we previously showed is dephosphorylated in a B55α-dependent manner12. It is important to note that our in vitro findings were observed in response to β-adrenergic receptor stimulation with isoproterenol, a setting of acute cardiac stress. While B55α was found to interact with HDAC5 in vehicle-treated cardiomyocytes in these experiments, this interaction increased ~3-fold in response to isoproterenol stimulation and was accompanied by a ~90% decrease in HDAC5 phosphorylation at Ser259. Our new findings suggest that PP2A-B55α is unlikely to be a major regulator of HDAC5 phosphorylation under basal conditions in vivo (i.e. in the absence of β-adrenergic stimulation) and may exert its effects on gene transcription via phospho-regulation of other proteins.
We found that cardiomyocyte-specific deletion of B55α in male hearts was associated with widespread changes in cardiac gene transcription (>500 differentially expressed genes), while the female cardiac transcriptome was largely unresponsive to B55α deletion (<10 differentially expressed genes). These data suggest that the PP2A-B55α holoenzyme may play a more dominant role in the male heart than the female heart under basal conditions, despite comparable B55α protein abundance. PP2A can bind to both the estrogen receptor66 and the androgen receptor67, but further work is required to investigate B subunit involvement in PP2A regulation of sex hormones. To date, the role of PP2A in the heart has been studied predominantly in male animals. Sex differences have not been evaluated in a setting of PP2A-C deletion or overexpression68,69,70,71. In mice with global deletion of B56α, there were no differences in LV dimensions or HW/TL ratio in males or females assessed at two months of age72. At six months of age, hearts of mice with homozygous knockout of B56α were smaller in males, but not in females, revealing a sex- and age-specific effect of this regulatory B subunit isoform on cardiac morphology72. Further work is required to investigate the relative abundance, subcellular localisation and activity of different PP2A holoenzymes in the male and female heart.
Hearts of male cardiomyocyte-specific B55α knockout mice had increased expression of extracellular matrix-related genes, leading to a small increase in LV collagen content, and decreased expression of genes involved in mitochondrial energy metabolism. Genes associated with the GO Cellular Component term ‘extracellular matrix’ (see Supp Table 8) included collagens (Col1a1, Col1a2, Col4a3, Col4a4, Col14a1), other ECM glycoproteins (Ahsg, Atrnl1, Cilp, Ecm1, Fbln1/2, Fn1, Hmcn2, Mfap4/5, Thbs3/4, Tnxb), metallopeptidases (Adamts2, Adamts10, Adamtsl2), regulators of ECM structure and organisation (Loxl2, Pcolce2, Ssc5d), small leucine-rich repeat proteoglycans (Aspn, Bgn, Hspg2, Prelp) and growth factors/signalling genes (Ccn2, Igfbp7, Ltbp3/4, Ndnf). Many of the proteins encoded by these genes (e.g. collagens, FN1, ADAMTS2, LTBP4) are secreted from fibroblasts, revealing a potential role for B55α in cardiomyocyte-fibroblast crosstalk. Cardiomyocytes serve an important paracrine function in regulating collagen production in fibroblasts73. The mechanisms by which B55α in male cardiomyocytes influences fibroblast gene transcription and extracellular matrix composition requires further investigation.
We detected a similar increase in LV collagen content in female cKO hearts, despite no differences in expression of Col1a1 or Col3a1, the predominant cardiac collagen isoforms. Expression of collagen IV isoforms (Col4a3 and Col4a4) tended to be higher in female cKO vs FC hearts, but these changes alone cannot account for the increase in LV collagen by picrosirius red staining given that male cKO mice displayed greater increases in Col4a3 and Col4a4, in addition to increased expression of several other collagen isoforms. It is possible that deletion of B55α in the female heart modulates the activity of enzymes involved in collagen degradation (e.g. TIMPs, MMPs, ADAMs), but investigation of this was beyond the scope of the current study. Of note, a previous study reported PP2A-dependent dephosphorylation of MMP-2 in isolated rat hearts subjected to ischemia/reperfusion, but the B subunit responsible for targeting PP2A catalytic activity to MMP-2 was not identified74. Further work is required to understand B55α-dependent sex differences in the transcriptional and post-translational regulation of cardiac collagen composition.
Pathway analysis of downregulated genes in hearts of male cardiomyocyte-specific B55α knockout mice revealed an enrichment of pathways associated with the mitochondria and oxidative phosphorylation (see Fig. 6d & Supp Table 8). A previous study reported differences in the expression of selected metabolic genes in hearts of mice with cardiomyocyte-specific deletion of the PP2A catalytic isoform PP2A-Cα between P1 and P11, but expression in the adult heart was not reported70. As FoxO1 has previously been identified as a key regulator of mitochondrial enzymes in the heart45, and B55α dephosphorylates FoxO1 in islet β-cells18, we hypothesised that differences in the expression of mitochondrial and metabolic genes in B55α knockout hearts may be due to phosphorylation-mediated inactivation of FoxO1. However, we observed no differences in the phosphorylation of FoxO1 at the pertinent phosphosites between genotypes, suggesting a FoxO1-independent mechanism by which B55α regulates the expression of metabolic genes. Further work is required to identify the signalling mechanisms by which B55α regulates gene transcription in cardiomyocytes.
Finally, our findings may have implications for the development of PP2A-targeting small molecules, which are promising cancer therapeutics. PP2A is inactivated in numerous solid and haematological tumours, and compounds that increase PP2A activity (e.g. FTY720) reduce tumour burden in preclinical models75. Conversely, PP2A inhibitors (e.g. LB100) have been shown to synergistically enhance the efficacy of other anticancer agents76. A major challenge for developing more effective cancer therapies is to identify the specific PP2A complexes to be targeted77. In this context, B55α and B56α target PP2A catalytic activity to different amino acid residues on the proto-oncogene c-Myc, with opposite effects on c-Myc stabilisation and oncogenic signalling43. PP2A-B55α-mediated dephosphorylation of Thr58 leads to c-Myc stabilisation and tumorigenesis, while PP2A-B56α-mediated dephosphorylation of Ser62 leads to c-Myc destabilisation and tumour suppression, revealing distinct roles for different PP2A holoenzymes43. PP2A B55 family members are emerging as important modulators of oncogenic signalling in numerous cancer settings78,79. B55α has been identified as a tumour suppressor in breast and prostate cancers, leukemias, and lung and thyroid carcinomas, and promotes tumour growth in pancreatic cancer settings79. Therapeutic approaches to activate or inhibit PP2A-B55α holoenzymes in specific cancer contexts need to consider the potential off-target effects of B55α modulation in the heart and other organs.
Our data show that partial loss of B55α in all cell types, or complete loss of B55α in cardiomyocytes, does not lead to pathological cardiac hypertrophy or dysfunction in mice in the absence of stress/injury. However, loss of B55α significantly altered the cardiac transcriptome in male mice and may predispose male hearts to pathology. Therefore, off-target cardiac effects of cancer therapeutics inhibiting B55α may be of concern in the male heart (and particularly in the aged male heart). Further research is required to investigate sex-specific responses to reduced B55α expression in settings of cardiac stress/injury, and if increasing B55α expression has any adverse effects on heart structure or function, or if it is cardioprotective.
In conclusion, this study provides the first characterisation of a PP2A B/B55 subfamily member in the mouse heart. Global disruption of the gene encoding B55α was embryonically lethal but did not adversely affect cardiac development. Loss of cardiomyocyte B55α did not impact overall heart size, atrial weight or LV dimensions in mice evaluated at 10–12 weeks of age. However, cardiomyocyte-specific deletion of B55α significantly altered the cardiac transcriptome in male mice, upregulating the expression of genes associated with the extracellular matrix and increasing LV collagen content, and downregulating the expression of genes associated with mitochondrial energy production. In contrast, few genes were differentially expressed in female hearts with and without cardiomyocyte B55α. In addition, mice with global heterozygous knockout of B55α had thinner LV walls and reduced mRNA, but not protein, expression of the lymphatic vessel marker Lyve1. These studies identify sex-specific functions for B55α in the heart under basal conditions and lay the foundation for future work investigating the role of B55α in settings of cardiac stress or injury.
Responses