Surface-hydroxylated single-atom catalyst with an isolated Co-O-Zn configuration achieves high selectivity in regulating active species
Introduction
SACs have captured substantial attention in water treatment due to their efficient metal atom utilization, high catalytic activity, and tunable electronic properties (Fig. 1a)1,2,3,4. Specifically, SACs are crucial for generating various active species necessary for diverse wastewater treatments. However, in real wastewater environments (particularly acidic ones), SACs encounter significant challenges. The unsaturated coordination environments of their active metal centers make them less stable compared to other heterogeneous catalysts. This instability often results in the leaching and diffusion of metal centers into the water, impairing catalytic performance and posing risks to the efficacy of water treatment5. For example, the degradation of p-hydroxybenzoic acid (HBA) by Fe SACs, as reported by Sun et al., was notably less effective under acidic conditions6. Additionally, synthesizing SACs usually involves complex and demanding techniques to achieve selective modulation of active species tailored for specific oxidant systems, limiting their applicability across different systems. For instance, Zhan and Zhang et al. developed SACs featuring CoN2+2 and CoN1O2 configurations, achieving nearly 100% selectivity for generating 1O2 and Co(IV) = O from activated PMS, respectively7,8. Yet, reports on adapting these SACs for other oxidant systems are lacking. Designing a versatile SAC that can effectively activate and selectively generate active species across various oxidant systems remains a largely unexplored area.

a Overview diagram of SACs. b Work function of different materials. c, d Work functions and corresponding structural models of (c) ZnO, (d) CoSAs-ZnO. e Comparison of the formation energies for different positions of Co substitution. f Schematic diagram of the synthesis process of CoSAs-ZnO (The first figure shows the precursor mixing. The second panel represents Co-modified Zn-MOF, and the third panel represents CoSAs-ZnO. The first arrow represents the synthesis step from the first panel to the second panel, and the second arrow represents the synthesis step from the second panel to the third panel). g Comparison of k-values with those of reported catalysts for SMX removal.
Furthermore, current SACs utilize carbon materials as supports that primarily function to anchor metal atoms, showing significant inertness and not actively participating in the catalytic reaction9. In complex wastewater environments, maintaining an appropriate pH often necessitates additional buffering agents, escalating treatment costs. Identifying an active support that can engage in the catalytic reaction and stabilize the reaction’s pH is crucial. Typically, the metal sites on single carbon material support exhibit symmetrical structures, limiting oxidant utilization and hindering the maximization of oxidant efficiency. Recent studies have shown that asymmetric coordination structures at metal sites can optimize the adsorption behavior of oxidants, effectively regulating interactions and electronic structures at metal sites during the catalytic process10. For instance, Zhang et al. demonstrated significant benefits in urea oxidation reactions using an atomically isolated asymmetric site structure with a Ni-O-Ti configuration over a symmetrical Ni-O-Ni setup11. Therefore, introducing asymmetric monoatomic sites may significantly enhance catalytic reactions.
Given these challenges, developing multifunctional support that not only anchors single atoms to create asymmetric sites but also actively participates in catalytic reactions and stabilizes reaction pH is essential. Metal oxides, favored for constructing SACs with asymmetric configurations, are promising in this regard12. Notably, amphiphilic metal oxides like zinc oxides can maintain the pH of the reaction system. For example, Xing et al. developed a Zn-Fe layered double hydroxide (ZnFe-LDH) that not only maintained a neutral pH microenvironment but also activated PMS to achieve 100% selective generation of Fe(IV) = O13. In addition, the amphoteric nature of zinc hydroxide has been used to create a neutral microenvironment, facilitating the self-stabilization of the reaction across a broad pH range and enhancing the degradation of contaminants14. In this study, ZnO, known for its biocompatibility and amphiphilic properties, is selected as the carrier for SACs. The synthesized ZnO has numerous hydroxyl functional groups on its surface, corroborating reports by Zheng and Xing et al.15,16. However, the specific roles of these groups in the reaction process require further investigation.
This study sets the stage for further exploration of amphiphilic ZnO as an active carrier to host highly active asymmetric transition metal single-atom sites for the activation of different oxidants to form specific active species. The remaining questions include the precise modulation of the asymmetric structure of SACs, the construction of highly active multifunctional carriers, the identification and quantification of active species, and a comprehensive evaluation of pollutant degradation mechanisms in systems dominated by different active species and the toxicity of their reaction products. These investigations will illuminate the intricate relationship between structure and catalytic performance in SACs and further the development of advanced catalytic water treatment technologies.
To address the aforementioned issues, this study strategically selected ZnO as the carrier material to develop and construct original SACs. Our research concentrated on the currently relevant asymmetric oxidants, PMS and PAA. Utilizing density functional theory (DFT) for precise predictions, we cleverly engineered an SAC featuring surface hydroxylation and an asymmetric Co-O-Zn configuration. This structure effectively activated both PMS and PAA, achieving remarkably high utilization efficiencies of 86% and 89%, respectively. This led to an exceptionally rapid degradation of sulfamethoxazole (SMX), with almost complete removal achieved within 6 min. The normalized kinetic constants measured were 586.7 and 427.8 min−1 M−1, significantly surpassing those reported in previous studies. Moreover, this SAC demonstrated impressive selectivity in PMS and PAA systems, achieving 96.4% and 95.1% selectivity for generating SO4•− and 1O2. In-depth DFT calculations highlighted that the surface hydroxyl groups on ZnO played a pivotal role in modulating the adsorption configurations of the two oxidants, thus enabling the selective generation of active species in each respective system. Considering the specific nature of the reactive species generated in the two systems, different treatment scenarios were used. In the SO4•−-dominated system, the focus was on treating electron-deficient benzoic acid wastewater, where pilot-scale experiments demonstrated a removal efficiency nearing 100% after multiple successive treatments. In the 1O2-dominated system, the methodology was applied to the conversion of high-value chemicals, achieving a conversion of benzyl alcohol to benzaldehyde with a selectivity of 89%. In addition, potential degradation pathways of SMX in both systems were thoroughly analyzed using DFT Fukui function analysis and liquid chromatography-mass spectrometry. The toxicity of the reaction systems was comprehensively evaluated using toxicity analysis software, alongside microbial, plant, and animal experiments. The environmental significance and viability of the CoSAs-ZnO/PMS and CoSAs-ZnO/PAA systems were quantitatively assessed through LCA. This study offers insights into the design of SACs and presents promising prospects for the treatment of wastewater and the conversion of high-value chemicals.
Results
Theoretical calculation predictions guiding the precise design of asymmetric Co SAs
To accurately construct asymmetric M-O-Zn sites, DFT predictions were employed. We initially performed theoretical calculations to determine the work function, which was defined as the minimum energy required to move an electron from the Fermi level to the vacuum17,18. This method was used to evaluate the effectiveness of various transition metals, including Fe, Co, Ni, and Cu. The results showed that the surface work functions for pristine ZnO, Fe, Cu, Ni, and Co embedded in ZnO were 6.12, 5.90, 5.84, 5.73, and 5.64 eV, respectively (Fig. 1b–d, Supplementary Fig. 1 and Supplementary Table 1). These findings indicate that anchoring these metal atoms into ZnO decreases the energy barrier for electron escape by lowering the surface vacuum level and raising the Fermi level, thereby enhancing oxidant activation. Of all the metals tested, Co demonstrated the lowest work function when incorporated into ZnO, suggesting it has the best electron-donating ability. Further, to identify the optimal substitution site for Co in ZnO, we developed models for various potential sites. The optimized models revealed that the formation energy was the lowest (Er = − 12.4 eV) when Co atoms replaced Zn and bonded with adjacent O atoms (Fig. 1e and Supplementary Fig. 2). This indicated that the Co-substituted Zn atom configuration was the most energetically favorable and structurally stable.
The synthesis of these catalysts was guided by these theoretical insights (Fig. 1f). The Co-modified Zn-MOF was synthesized by a self-assembly method using 2-methylimidazole as an organic linker, followed by one-step calcination. This process successfully produced CoSAs-ZnO catalysts with an asymmetric Co-O-Zn configuration. To validate the successful construction of this specific configuration, normalized k-values were compared with those of previously reported efficient catalysts (Fig. 1g). The results showed that the asymmetric catalyst systems achieved an ultra-high rate of degradation of SMX with normalized k-values of 586.7 for the PMS system and 427.8 min−1 M−1 for the PAA system, both of which are substantially higher than previously reported values. This preliminary confirmation of the successful construction of SACs with an asymmetric Co-O-Zn configuration indicates significant potential for application in wastewater treatment.
Validation of atomically isolated asymmetric Co-O-Zn site in Co SAs
To verify the successful construction of the Co-O-Zn asymmetric configuration in the SAC, comprehensive characterizations were performed. X-ray diffraction (XRD) spectra (Fig. 2a) confirmed that both ZnO and CoSAs-ZnO maintained the hexagonal wurtzite structure characteristic of zincite (JCPDS No. 36-1451), with no evidence of secondary phases19. This finding highlighted the high crystallinity of ZnO and the atomic level dispersion of Co within the ZnO matrix20. The doping of Co single-atom sites resulted in notable shifts of the (100) and (101) plane diffraction peaks to higher 2θ angles, indicative of lattice extension. The similarities in valence, electronic configuration, and ionic radius between Co2+ and Zn2+ suggested that Co might substitute Zn in the lattice, potentially forming an asymmetric Co-O-Zn structure15,21. Fourier-transform infrared spectroscopy (FTIR) provided further insight into the chemical composition and structural changes in the CoSAs-ZnO catalyst (Fig. 2b). The peak at 449 cm−1, corresponding to Zn-O stretching vibrations, shifted to 439cm−1 after Co incorporation. This shift was attributed to the interaction between Co-O and Zn-O vibrations, induced by the presence of Co atoms15. In addition, 1634 cm−1 and 3449 cm−1, associated with O-H stretching vibrations, highlighted the presence of numerous hydroxyl groups in the ZnO structure16. The role of these hydroxyl groups in the catalytic process will be explored in the subsequent sections on theoretical calculations.
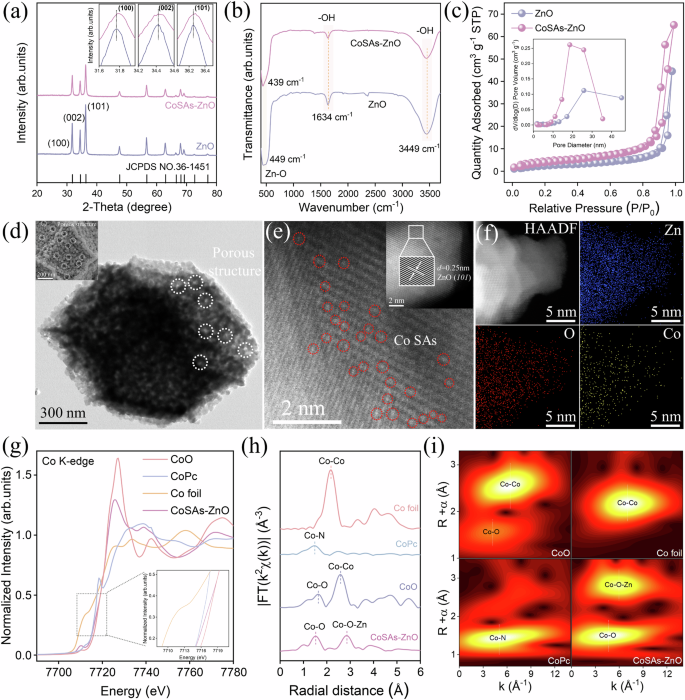
a XRD pattern of ZnO and CoSAs-ZnO (inset: enlarged view of (100), (002), and (101) crystal facets). b FTIR spectra of ZnO and CoSAs-ZnO. c Nitrogen adsorption-desorption isotherm of ZnO and CoSAs-ZnO (inset: the corresponding pore diameter distribution). d TEM image of CoSAs-ZnO (inset: SEM image of CoSAs-ZnO). e HAADF-STEM image of CoSAs-ZnO (single atoms are marked by red circles; inset: HRTEM image of CoSAs-ZnO). f HAADF-STEM elemental mappings of CoSAs-ZnO with original, Zn, O, and Co. g XANES spectra of CoSAs-ZnO at the Co K-edge (inset: magnification of local areas). h FT k3-weighted EXAFS spectra of CoSAs-ZnO and references. i WT-EXAFS for K-edge for Co foil, CoO, CoPc, and CoSAs-ZnO.
Microscopic analyses using scanning electron microscopy (SEM) and transmission electron microscopy (TEM) further elucidated the physical form of Co on the CoSAs-ZnO surface. ZnO displayed an orthododecahedral morphology with a rough and porous surface. Elemental mapping confirmed the uniform distribution of Zn and O elements (Supplementary Figs. 4–7). After Co incorporation, the morphology of CoSAs-ZnO remained largely unchanged, with uniform distribution of Zn, O, and Co elements maintained (Fig. 2d and Supplementary Figs. 8–11). To further compare the structural changes post-Co introduction, N2 adsorption-desorption analysis was utilized. This analysis revealed that CoSAs-ZnO had a higher surface area and more optimal pore structure (Fig. 2c and Supplementary Table 2). These structural enhancements were likely responsible for the improved capacity of the catalyst to adsorb and concentrate oxidants and pollutants, thereby enhancing its catalytic performance.
To robustly validate the successful construction of the asymmetric Co-O-Zn structure in CoSAs-ZnO, a comprehensive analysis was conducted using different advanced characterization techniques, including aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM), X-ray photoelectron spectra (XPS), and X-ray absorption fine structure (XAFS) analyses. HAADF-STEM images of CoSAs-ZnO (Fig. 2e and Supplementary Fig. 12) displayed numerous bright spots encircled in red, indicating the presence of highly dispersed Co sites. The accompanying elemental mapping (Fig. 2f and Supplementary Fig. 13) confirmed the uniform distribution of Zn, O, and Co throughout the CoSAs-ZnO structure. In addition, measurements of the lattice spacing of CoSAs-ZnO, recorded at 0.25 nm, correspond to the (101) crystalline plane of ZnO22. XPS was utilized to further characterize the elemental valence states. The high-resolution XPS spectra for Co 2p displayed peaks at 781.2 eV and 786.0 eV, which are indicative of the Co 2p3/2 orbitals of Co2+ and the satellite peaks of Co2+, respectively (Supplementary Fig. 14)23,24. Notably, no peaks corresponding to Co clusters or Co oxides were observed. In addition, high-resolution XPS spectra for O 1 s exhibited peaks at 530.3 eV and 531.7 eV (Supplementary Fig. 16), attributable to Zn-O and Zn-OH bonds, respectively25. This corroborated the presence of abundant hydroxyl groups on the ZnO surface, aligning with previous FTIR results.
To further corroborate the existence of the Co-O-Zn configuration in CoSAs-ZnO, extensive XAFS analyses were performed (Supplementary Fig. 17 and Supplementary Table 3). The X-ray absorption near edge structure (XANES) spectrum (Fig. 2g) of the Co K-edge indicated that the absorption edge of Co atoms in CoSAs-ZnO closely resembled that of CoO, suggesting that the Co atoms predominantly exist in a + 2 valence state, which was consistent with the findings from XPS analyses12. Fourier transform extended X-ray absorption fine structure (FT-EXAFS) analysis (Fig. 2h) revealed two distinct peaks at 1.5 Å and 2.8 Å corresponding to Co-O and Co-O-Zn coordinations, respectively. Importantly, the absence of Co-Co peaks in these spectra confirmed the lack of Co clusters, affirming the atomic dispersion of Co within the structure26. Wavelet transforms (WT)-EXAFS results (Fig. 2i) further delineated two distinct peaks at 4.7 Å and 5.9 Å attributed to Co-O and Co-O-Zn shells, respectively, in CoSAs-ZnO. The absence of Co-Co peaks reiterated the atomically dispersed nature of Co sites26.
Together, these comprehensive findings verified that Co atoms replaced Zn atoms in the ZnO lattice, resulting in the formation of an atomically isolated asymmetric Co-O-Zn structure. This configuration aligned well with DFT predictions, which indicated the lowest formation energy for this specific substitution, reinforcing the successful integration of Co within the ZnO matrix to enhance catalytic functionality.
Comprehensive assessment of catalytic activity and stability in Fenton-like systems
The characterization results robustly confirmed the successful creation of SACs with an asymmetric Co-O-Zn structure. To evaluate whether this catalyst could be applied across different oxidizing systems, we selected two prevalent and asymmetric oxidants, PMS and PAA, and performed the subsequent related studies. The optimized concentrations of CoSAs-ZnO, SMX, PMS, and PAA were established (Supplementary Figs. 18–25 and Supplementary Tables 4, 5). Unless specified, experimental conditions in subsequent studies remained consistent. Comparative experiments demonstrated negligible adsorption of SMX by both CoSAs-ZnO and ZnO (Fig. 3a and Supplementary Table 6). Similarly, the use of PMS and PAA alone had minimal impact on SMX removal (Supplementary Fig. 26 and Supplementary Table 6). Notably, the CoSAs-ZnO catalyst exhibited outstanding ability to activate PMS and PAA, achieving complete removal of SMX within 6 min (Fig. 3a). In contrast, the ZnO/PMS and ZnO/PAA systems achieved only a 5% SMX removal. When compared to the ZnO/PAA system, the CoSAs-ZnO/PAA system exhibited a 13-fold increase in degradation rate and a 55-fold increase in the kinetic constant (Fig. 3b). These results demonstrated the efficacy of the CoSAs-ZnO catalyst with a Co-O-Zn structure in activating asymmetric oxidants, facilitating the generation of highly oxidative species (Supplementary Fig. 27).

a Degradation efficiencies of SMX in different reaction systems. (Conditions: [Catal.]0 = 0.1 g L−1, [PMS]0 = 0.15 mM, [PAA]0 = 0.18 mM, [pH]0 = 6.3, [SMX]0 = 10 mg L−1, [Temp.]= 20 ± 2 °C). b Comparison of degradation efficiencies and reaction rate constants of different reaction systems. c Comparison utilization efficiencies of PMS and PAA by CoSAs-ZnO. d Evaluation of SMX removal using CoSAs-ZnO/PMS and CoSAs-ZnO/PAA systems with six cycles. e pH changes of CoSAs-ZnO/PMS and CoSAs-ZnO/PAA reaction systems under different initial pH conditions. f Degradation efficiencies of SMX by CoSAs-ZnO/PMS and CoSAs-ZnO/PAA systems at different initial pH. g TOC removal efficiencies by different reaction systems. h Removal of multiple pollutants by CoSAs-ZnO/PMS and CoSAs-ZnO/PAA reaction systems. i Effect of background ions on the efficiency of SMX degradation by CoSAs-ZnO/PMS and CoSAs-ZnO/PAA reaction systems. The error bars represent the standard deviation of three replicate tests.
To thoroughly assess the effectiveness of the CoSAs-ZnO catalysts with the Co-O-Zn asymmetric configuration in activating asymmetric oxidants, the consumption of oxidants was meticulously quantified. Within 6 min, 86% of PAA was utilized by the CoSAs-ZnO catalyst (Fig. 3c), indicating that the atomically isolated asymmetric Co-O-Zn sites possessed a high capability for activating the oxidant. In addition, the specially designed CoSAs-ZnO, featuring the Co-O-Zn asymmetric structure, demonstrated remarkable stability, maintaining approximately 98% removal of SMX by activated PAA even after six reaction cycles (Fig. 3d). Furthermore, the amphoteric nature of ZnO significantly contributed to maintaining a neutral microenvironment within the reaction system, enhancing stability (Supplementary Fig. 28). Real-time pH monitoring with and without the catalyst across various initial pH values confirmed that the CoSAs-ZnO system could sustain a nearly neutral pH (~ 6.0) regardless of the initial acidic or alkaline conditions (Fig. 3e and Supplementary Figs. 29, 30). This self-sustaining pH effect was crucial to the system’s performance over a wide pH range of 3.0 to 9.0 (Fig. 3f and Supplementary Tables 7, 8), facilitating the practical treatment of complex wastewater scenarios and the reduction of leaching of metal Co (Supplementary Fig. 31). The PAA-activated system also showed significant improvements in the removal rate of total organic carbon (TOC), achieving up to 57.2% mineralization of SMX within 6 min, a substantial enhancement over the 10.7% efficiency of the ZnO/PAA system (Fig. 3g). This system proved effective in degrading a variety of pollutants, including rhodamine B (RhB), methylene blue (MB), sulfadiazine (SDZ), sulfathiazole (STZ), and sulfapyridine (SPY), nearly eliminating all within 6 min (Fig. 3h, Supplementary Figs. 32, 33 and Supplementary Tables 9, 10). In addition, the PAA-activated system demonstrated strong resistance to external interference, maintaining robust performance in the presence of various inorganic ions and organic compounds (Fig. 3i, Supplementary Figs. 38–41 and Supplementary Table 12). Similarly, the constructed CoSAs-ZnO/PMS system with atomically isolated asymmetric Co-O-Zn sites also possessed high efficiency in water purification and held significant potential for practical applications (Fig. 3i, Supplementary Figs. 34–37 and Supplementary Table 11).
In conclusion, both the PAA and PMS activation systems using CoSAs-ZnO exhibited exceptional performance, efficient oxidant utilization, impressive stability, and robust environmental resistance. Although conventional wisdom typically suggests that oxidant systems of the same type produce similar active species, the outstanding performance of both PMS and PAA activation systems raises questions about the similarity of their active species. This area remains to be explored further.
Exclusivity of highly active species in different asymmetric oxidant activation
To explore the differences in active species across various oxidant-activated systems of Co-O-Zn configurations in Co SACs, several scavengers were employed: tert-butyl alcohol (TBA) for hydroxyl radicals (•OH), methanol (MeOH) for sulfate radicals (SO4•−), 2,4-HD for organic radicals (R-O•), TEMPOL for superoxide radicals (•O2–), and L-histidine for singlet oxygen (1O2)27,28,29,30. In the CoSAs-ZnO/PAA system, the SMX degradation efficiencies were minimally affected or only weakly inhibited by the introduction of TBA and 2,4-HD, initially suggesting that •OH and R-O• were not the primary active species in this system (Fig. 4a and Supplementary Fig. 42). However, significant inhibition of degradation efficiency was observed with the addition of TEMPOL and L-histidine, indicating that •O2– and 1O2 might be present in the CoSAs-ZnO/PAA system.

a Degradation of SMX in the presence of various scavengers in the CoSAs-ZnO/PMS and CoSAs-ZnO/PAA reaction systems. b DMPO spin-trapping ESR spectra for DMPO-SO4•− in the CoSAs-ZnO/PMS system. c TEMP-based ESR spectra for 1O2 in the CoSAs-ZnO/PAA system. d Steady-state concentrations of active species in different reaction systems. e The proportion of active species in the CoSAs-ZnO/PMS and CoSAs-ZnO/PAA system. f Cyclic voltammetric curves of ZnO and CoSAs-ZnO electrodes (inset: LSV curves of ZnO and CoSAs-ZnO electrodes). g EIS spectra of ZnO and CoSAs-ZnO. h Amperometric i-t curve measurements upon the addition of PMS, PAA and SMX using ZnO and CoSAs-ZnO as the working electrode. i In-situ Raman spectra of different reaction systems. j Effect of pre-mixing time on SMX removal in the CoSAs-ZnO/PMS and CoSAs-ZnO/PAA reaction systems. The error bars represent the standard deviation of three replicate tests. k The proposed reaction mechanisms in the CoSAs-ZnO/PMS and CoSAs-ZnO/PAA systems.
Electron spin resonance (ESR) measurements further supported these findings. Notably, no •OH and R-O• signals were detected in the ESR spectra of the CoSAs-ZnO/PAA system, confirming that neither of these species was the main active species (Supplementary Fig. 44). R-O• was additionally tested using TEMPO, a known organic radical trapping agent31. The mass spectra (Supplementary Fig. 46) did not show signals for the TEMPO-CH3 adduct (m/z = 172.1697) or the TEMPO-OOCCH3 adduct (m/z = 216.1595), providing strong evidence that methyl (-CH3) and acetoxy (CH3COO•) radicals were not present in the system. Furthermore, experiments demonstrated that Mn2+ quenched acetyl peroxyl radicals without depleting PAA32. The addition of Mn2+ had minimal impact on the reaction rate, further validating the absence of acetyl (per)oxygen radicals (Supplementary Fig. 47). However, characteristic signals for DMPO-•O2– and TEMP-1O2 were observed in the ESR spectra of the CoSAs-ZnO/PAA system (Fig. 4c and Supplementary Fig. 44). To accurately confirm the presence of •O2– and 1O2, experimental studies were conducted using NBT and DPBF-specific probes18,33. The results indicated that •O2– was absent in the PAA-activated system (Supplementary Fig. 48). Notably, the PAA-activated system showed a gradual decrease in the absorption peak of DPBF near 410 nm as the reaction progressed, strongly confirming the generation of 1O2 (Supplementary Fig. 50). This analysis demonstrated that the primary active species in the PAA activation system was indeed 1O2. Following the same line of research, the active species in the PMS activation system were accurately identified (Fig. 4b, Supplementary Figs. 43, 45, 49–51). Similarly, investigations into the PMS activation system also confirmed that the primary active species was SO4•−34.
To further quantify the contributions of 1O2 and SO4•− in the PAA and PMS systems, respectively, computational quantification was also performed using benzoic acid (BA), nitrobenzene (NB), and furfuryl alcohol (FFA) as probes (Fig. 4d and Supplementary Figs. 52, 53)35,36. According to the calculations based on Equations. S1-S22, the PAA-activated system exhibited the highest steady-state concentration of 1O2, accounting for up to 95% of the reaction. Conversely, the PMS-activated system showed the highest steady-state concentration of SO4•−, with 1O2 contributing negligibly. The contribution of SO4•− to the reaction was as high as 96.4% (Fig. 4e). In conclusion, these findings illustrate that similar oxidant systems can operate via radically different active species pathways, one dominated by radicals and the other by non-radicals. The results indicated the limitations of relying solely on individual validation methods, which could lead to misinterpretations. This highlights the critical need for employing a comprehensive approach that combines multiple analytical techniques to accurately assess both the formation and activity of reactive species in catalytic systems.
To further elucidate the intrinsic catalytic activity and electron transfer pathways of SACs with asymmetric Co-O-Zn configurations, a series of electrochemical experiments were conducted. Through cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) analyses, it was revealed that CoSAs-ZnO exhibited higher specific capacitance and lower charge transfer resistance (Fig. 4f, g), confirming that the asymmetric Co-O-Zn site enhanced electron transfer efficiency37,38. Linear sweep voltammetry (LSV) further indicated that the CoSAs-ZnO electrode had a significantly higher current density compared to standard ZnO (Fig. 4f), signifying more rapid electron transfer39. Chronoamperometry studies provided i-t curves (Fig. 4h) that offered insights into electron transfer pathways40,41. Dosing PMS or PAA led to a noticeable response in current density for CoSAs-ZnO, indicative of electron redistribution caused by interactions between these oxidants and the catalytic sites. In contrast, the addition of SMX caused minimal changes, underscoring the specific roles of PMS and PAA in forming active complexes for generating SO4•− and 1O2, respectively. Further analysis involved open-circuit potential monitoring and pre-mixing experiments. An immediate increase in potential on the CoSAs-ZnO electrode upon adding PMS or PAA was observed (Supplementary Fig. 54), suggesting the formation of active complexes on the catalyst surface42. The lack of a potential decrease after SMX addition ruled out the involvement of catalyst-mediated electron transfer processes. Pre-mixing experiments, where CoSAs-ZnO was combined with PMS or PAA before adding SMX, displayed a reduction in SMX removal efficiency with longer pre-mixing times (Fig. 4j and Supplementary Figs. 55, 56). This supported the hypothesis that PMS and PAA rapidly decomposed to generate SO4•− and 1O2 in the absence of SMX, confirming that ETP mechanisms were not involved43. In situ Raman spectroscopy tracked the adsorption and evolution of PAA and PMS on the CoSAs-ZnO surface (Fig. 4i). In the PAA-activated system, the characteristic peak at 875 cm−1 of PAA was significantly weakened upon the introduction of CoSAs-ZnO44, indicating efficient utilization of PAA and subsequent 1O2 generation. In the PMS-activated system, peaks at 882 and 1060 cm−1, likely associated with HSO5– in PMS, and a peak at 980 cm−1 corresponding to S = O bond stretching vibrations in SO42− were noted45. After introducing CoSAs-ZnO, these characteristic peaks were markedly reduced, demonstrating efficient PMS utilization. A significant shift of the peak at 1060 cm−1 suggested a strong interaction between PMS and active sites on CoSAs-ZnO46,47. In addition, a new peak at 834 cm−1 indicated the formation of active peroxides (PMS*), contributing to the generation of SO4•−. These observations collectively demonstrated that electron transfer from CoSAs-ZnO to PMS and PAA favored the targeted generation of SO4•− and 1O2. This conclusion aligned with the results from active species quantification and other electrochemical experiments, highlighting the catalyst’s Co-O-Zn asymmetric structure in enhancing the efficient utilization of oxidants and selectively generating active species.
Overall, the results showcased the successful development of Co SACs with Co-O-Zn structures (Fig. 4k). These catalysts are capable of selectively generating a single active species across various oxidant systems, illustrating their versatility in activating multiple oxidants. This distinctive structural design enables efficient and robust reactions, making these catalysts adaptable for targeted and flexible applications in different wastewater treatment scenarios.
Insight into the mechanisms of selective high-activity species production
To gain insight into the mechanism of highly selective active species generation within SACs with Co-O-Zn asymmetric configurations, theoretical calculations were performed. Initial investigations focused on the incorporation of single Co atoms into the ZnO surface, examining charge density distribution and active site exposure. The pristine ZnO surface exhibited a well-defined charge density distribution and electrostatic potential symmetry (Fig. 5a, b). However, upon substituting Zn with Co atoms, a significant alteration in the electrostatic distribution was observed. This substitution enhanced the electropositivity of the Co atoms, thereby strengthening their interaction with oxygen atoms in oxidant molecules. Moreover, surface -OH groups displayed pronounced positive electrostatic properties, suggesting that adsorption was primarily driven by van der Waals forces. Further studies indicated that the adsorption of PAA on both CoSAs-ZnO and ZnO surfaces was predominantly governed by hydrogen bonding, a fundamentally strong van der Waals force. Spin charge analysis (Fig. 5c) further revealed that introducing Co atoms induced substantial spin polarization at the Co site and neighboring oxygen atoms. This polarization facilitated the transfer or acceptance of unpaired electrons, achieving a lower energy state. Notably, the Co sites exhibited a strong electron-donating tendency, contributing significantly to oxidant activation. These changes in electronic structure and spin dynamics were corroborated by density of states (DOS) analysis, highlighting the profound impact of Co incorporation on the catalytic properties of ZnO.

a Electrostatic potentials of ZnO and CoSAs-ZnO surfaces (blue means negatively charged, red means positively charged). b Charge density distributions of ZnO and CoSAs-ZnO surface. c Spin charge densities of ZnO and CoSAs-ZnO (the orange area represents the spin-charge). d pDOS of ZnO, CoSAs-ZnO, PMS and PAA. e PMS and PAA adsorption energies, differential charge density, and Bader charge analysis of ZnO and CoSAs-ZnO, respectively (The pink and green areas represent regions of electron loss and gain, respectively). f Calculated Gibbs free energy for PMS activation on CoSAs-ZnO. g Reaction steps for the activation of PMS by CoSAs-ZnO to generate different active species. h Calculated Gibbs free energy for PAA activation on CoSAs-ZnO. i Reaction steps for the activation of PAA by CoSAs-ZnO to generate different active species.
The incorporation of Co atoms induced spin polarization on the exposed ZnO surface, meanwhile influencing oxygen atom adjacent to Co atom (Supplementary Fig. 57). Specifically, the β-spin projected density of states (PDOS) for oxygen (O) shifted upward, approaching the Fermi level, as depicted in Supplementary Fig. 58 and Supplementary Fig. 60. This shift resulted in an elevation of antibonding orbitals and a decrease in electron occupancy, potentially enhancing the adsorption of oxidants if O atoms served as adsorption sites. Concurrently, alterations in the surface hydrogen atoms were found to be negligible, as illustrated in Supplementary Fig. 61. For Zn atoms, the d-band PDOS were predominantly localized around − 6.0 eV, with a lower d-band center at − 6.11 eV, suggesting a reduced reactivity of Zn towards PMS adsorption. In contrast, the α-spin d-band PDOS of Co atoms displayed a distribution ranging from − 6.0 eV to the Fermi level, with a d-band center established at − 4.05 eV. This distribution, coupled with the observed better overlap of PDOS with oxygen, indicates a more effective p-d orbital hybridization between the α-spin orbitals of Co and the oxygen in PMS. This hybridization suggests a stronger interaction between Co and PMS, which could be pivotal for the catalytic activity of the material (Fig. 5d and Supplementary Fig. 59). Furthermore, the β-spin d-band electrons were positioned at a higher energy level, elevating the d-band center to 0.34 eV and lifting the antibonding orbitals upon oxidant adsorption, thus reducing electron occupancy in these orbitals. This intricate interplay of electronic properties significantly enhanced the material’s catalytic capabilities, particularly in oxidant activation and selective species generation.
Further investigations into the adsorption energy and Bader charge transfer for PMS and PAA on both ZnO and CoSAs-ZnO surfaces demonstrated that the introduction of Co did not significantly enhance the adsorption energy or charge transfer for either PMS or PAA (Fig. 5e and Supplementary Figs. 62–65). However, the presence of Co optimized the adsorption configurations for PMS, shifting from predominantly hydrogen-bonding interactions to Co-O bond interactions, which were crucial for the selective generation of SO4•−. In the CoSAs-ZnO/PAA system, the Co-induced electronic structure modifications were insufficient to overcome the inherent steric hindrance, resulting in PAA adsorption being predominantly governed by van der Waals forces and maintaining an unaltered adsorption configuration. Nevertheless, the presence of Co proved essential for the selective generation of 1O2, as evidenced by Gibb’s free energy calculations.
To gain deeper thermodynamic insights into the catalytic process, the Gibbs free energy associated with the generation of active species in both PMS and PAA-activated systems was calculated. In the PMS-activated system, the rate-determining steps for the CoSAs-ZnO catalyst to produce SO4•− and 1O2 were identified as product desorption ((*SO4•− + *H2O→*H2O + SO4•−) and (*1O2 + *2HSO4–→*1O2 + *HSO4–)), with free energies of + 1.04 eV and + 1.41 eV, respectively. This indicated that the generation of SO4•- was thermodynamically more favorable than the production of 1O2 in this system (Fig. 5f, g and Supplementary Figs. 66, 67). In the PAA-activated system, the free energies associated with the formation of CH3COO• and 1O2 were compared (Fig. 5h, i and Supplementary Figs. 68–70). The results revealed that the energy required for the rate-determining step to produce 1O2 (*1O2→1O2, +0.52 eV) was significantly lower than that needed for CH3COO• generation (*CH2COO∙ + *H2O→ *H2O + CH3COO•, + 0.72 eV). This suggested that the formation of 1O2 was more thermodynamically feasible than that of CH3COO•. These findings explain the highly selective generation of SO4•− and 1O2 in the PMS and PAA-activated systems, respectively.
Theoretical calculations further underscored the crucial role of hydroxyl groups in regulating the adsorption configurations of PMS and PAA and optimizing the subsequent Gibbs free energy evolution. These calculations lead to the highly selective generation of SO4•− and 1O2 in the PMS and PAA-activated systems. This detailed approach elucidated the complex interactions between the catalyst’s structural properties and catalytic behavior and also provided a framework for optimizing catalyst models to better align with experimental designs.
Pilot-scale application evaluation and multi-scenario simulation
To assess the applicability of the constructed PMS and PAA-activated systems, a continuous flow reactor was established to degrade organic pollutants continuously (Fig. 6a). The CoSAs-ZnO catalytic membrane, prepared using vacuum-assisted technology, was initially tested using RhB to visually demonstrate the system’s effectiveness. During a 12 h operational period, the catalytic membrane system successfully removed nearly all of the RhB (Fig. 6b). Similarly, the removal efficiencies for SMX by the PMS and PAA-activated systems remained consistently around 99% over the same period (Fig. 6c). To further verify practical application feasibility, pilot-scale experiments were conducted to treat SMX wastewater. As shown in Fig. 6d, the photographs depicted the entire degradation setup used for this experiment. Specifically, 50 L of SMX wastewater was introduced into a 40 cm × 40 cm × 38 cm transparent Plexiglas tank and stirred using a mechanical stirrer. A polyurethane sponge was chosen as the carrier for catalyst immobilization. SEM images and elemental mapping results indicated that the sponge featured a dense skeletal structure with pores, and the CoSAs-ZnO catalysts were uniformly distributed across its surface (Fig. 6d and Supplementary Figs. 71–73). This arrangement allowed the sponge to remain suspended in the wastewater, enhancing the interaction between the catalyst, oxidant, and contaminants. In addition, the sponge design facilitated the recovery and reuse of the catalyst post-degradation, reducing costs and minimizing the risk of secondary contamination. These systems consistently delivered excellent treatment outcomes, with antibiotic removal efficiency reaching nearly 100% even after six cycles (Fig. 6e).

a Photograph of the continuous flow reactor. b Continuous removal of RhB by the CoSAs-ZnO/PMS and CoSAs-ZnO/PAA systems. c CoSAs-ZnO/PMS and CoSAs-ZnO/PAA systems for continuous removal of SMX. d Pilot-scale experiment diagram for treating 50 L of antibiotic wastewater (inset: Photographs of the original sponge and the sponge after loading with catalysts). e Six consecutive treatments of 50 L of SMX-contained wastewater in CoSAs-ZnO/PMS and CoSAs-ZnO/PAA systems. f Six consecutive treatments of 10 L of BA-contained wastewater in CoSAs-ZnO/PMS system. g Comparison of the selectivity of the ZnO/PAA and CoSAs-ZnO/PAA systems for the conversion of benzyl alcohol to benzaldehyde. h Comparison of the performance of the CoSAs-ZnO/PMS and CoSAs-ZnO/PAA systems for the degradation of BA-containing wastewater. i Reaction mechanisms for CoSAs-ZnO/PMS and CoSAs-ZnO/PAA systems and their suitability for different scenarios. j Relative environmental impact of environmentally relevant descriptors for treating 50 mL SMX (10 mg L−1) in Fenton-like processes with ZnO/PMS, Co3O4/PMS, and CoSAs-ZnO/PMS systems (1. Global warming; 2. Stratospheric ozone depletion; 3. Ionizing radiation; 4. Ozone formation, Human health; 5. Fine particulate matter formation; 6. Ozone formation, Terrestrial ecosystems; 7. Terrestrial acidification; 8. Freshwater eutrophication; 9. Marine eutrophication; 10. Terrestrial ecotoxicity; 11. Freshwater ecotoxicity; 12. Marine ecotoxicity; 13. Human carcinogenic toxicity; 14. Human non-carcinogenic toxicity; 15. Land use; 16. Mineral resource scarcity; 17. Fossil resource scarcity; 18. Water consumption).
To maximize the specific advantages of the active species generated in the PMS and PAA systems, strategic applications were considered for different scenarios. The PMS-activated system, known for its selective generation of SO4•− (Fig. 6f), was applied to electron-deficient recalcitrant BA wastewater, yielding 100% removal after six consecutive treatments, showcasing its distinctive advantage. Conversely, the highly selective production of 1O2 in the PAA-activated system was suited for high-value chemical synthesis. This capability was demonstrated in the selective oxidation of benzyl alcohol to benzaldehyde, with the CoSAs-ZnO/PAA system achieving a conversion selectivity of 89%, significantly outperforming the ZnO/PAA system which only achieved about 42% selectivity (Fig. 6g). In addition, to highlight the specificity of the PMS and PAA-activated systems, the removal of BA using both systems was separately assessed. The PAA-activated system managed only a 19% removal efficiency, which marginally increased to 25% with extended reaction times (Fig. 6h and Supplementary Fig. 74). In stark contrast, the PMS-activated system accomplished ultra-fast degradation of BA, achieving complete removal in less than 2 min. These findings illustrated that the specificities of the distinct activated species make them suitable for targeted scenarios, enabling efficient and precise treatment of specific wastewater types. In summary, these findings underscored the specific practical utility of the designed systems (Fig. 6i). The CoSAs-ZnO/PMS system was highly effective for treating electron-deficient recalcitrant pollutants due to its robust generation of SO4•−. Meanwhile, the CoSAs-ZnO/PAA system demonstrated potential for synthesizing high-value chemicals, attributed to its superior 1O2 generation. This tailored application of each system highlights their adaptability and effectiveness in addressing varied environmental challenges.
Life cycle assessment (LCA) was utilized to quantify the sustainability and environmental impact of the CoSAs-ZnO/PMS and CoSAs-ZnO/PAA processes48,49. For comparison, catalytic systems based on ZIF8-derived ZnO and ZIF67-derived Co3O4 catalysts were employed (Supplementary Figs. 75–77 and Supplementary Tables 19–24). The analysis encompassed the entire water treatment process, including catalyst preparation and pollutant degradation, using 18 relevant descriptors, such as global warming potential and freshwater/marine ecotoxicity (Fig. 6j and Supplementary Figs. 78–80). The results indicate that the synthesized CoSAs-ZnO exhibits significant advantages over both ZnO and Co3O4, as reflected in its lower impact scores across most descriptors. This evidence suggests that the CoSAs-ZnO synthesized in this study is more cost-effective and environmentally friendly.
Contaminant removal pathway analysis and systematic real toxicity assessment
To elucidate the SMX degradation pathways within SACs featuring Co-O-Zn asymmetric configurations, DFT calculations were employed to identify the most vulnerable sites on the (Fig. 7a, b and Supplementary Figs. 81, 82, Supplementary Table 14). Orbital-weighted Fukui functions, specifically f0 and f–, were utilized to analyze SMX’s structural reactivity (Fig. 7b and Supplementary Fig. 82)50,51. This analysis revealed that the nitrogen atom at the 7(N) position was most susceptible to attack by both SO4•− and 1O2, as it exhibited the highest f0 and f– values52,53. Additionally, the sulfur atom at the 1(S) position showed elevated f0 values, indicating its vulnerability to SO4•- attack. Molecular electrostatic potential analysis (Supplementary Fig. 81) further highlighted that the aniline region of SMX possessed high positive charge values, suggesting its reactivity towards attacking species53. Moreover, the highest occupied molecular orbital (HOMO) of SMX (Fig. 7a) was predominantly situated on the benzene ring, reinforcing its susceptibility to attack. Consequently, both the 7(N) and 12(C) positions were identified as the most vulnerable sites in the two catalytic systems, while the 1(S) position was particularly susceptible to attack within the CoSAs-ZnO/PMS system.

a HOMO, and LUMO of SMX molecule. b Calculated the Fukui index of SMX. c Analysis of possible degradation pathways of SMX in the CoSAs-ZnO/PMS and CoSAs-ZnO/PAA systems. d Mortality ratio of wheat seeds cultured in the solution treated with different systems. e Violin plots of wheat shoot length distribution in various systems. f Photographs of E. coli colonies cultured in solution treated with different systems. g, Growth of zebrafish cultured in the solution treated with different systems (PE: pericardial edema, SC: spinal curvature, TM: tail curvature).
The degradation pathways of SMX in the two catalytic systems were further elucidated by integrating results from liquid chromatography-mass spectrometry with DFT findings (Fig. 7c, Supplementary Figs. 83, 84 and Supplementary Table 15). In the PMS-activated system, the -NH2 group on the benzene ring of SMX was first oxidized to form P1 (m/z = 283)54. This intermediate then underwent cleavage of the oxazole ring and double bond, resulting in P2 (m/z = 262), which was further oxidized to produce P3 (m/z = 218) and P4 (m/z = 142)55. In the PAA-dominated system, SMX underwent deamination and C-N bond cleavage due to the attack of 1O2 to produce P5 (m/z = 238) and P6 (m/z = 157). An alternative pathway involved the conversion of SMX to P7 (m/z = 270), which then underwent oxidation of the -NH2 group to produce P8 (m/z = 284), and the S-N bond in P8 was broken to form P9 (m/z = 98). Ultimately, these intermediates were mineralized into water and carbon dioxide through further oxidation processes.
To assess the practical applicability of these catalytic systems, a preliminary estimation of the toxicity of SMX and its intermediates was conducted using analytical software (Supplementary Figs. 85, 86, and Supplementary Table 16). The findings indicated that the toxicity levels of the degradation products were nearly non-toxic for both PMS and PAA-activated systems. To accurately assess the true toxicity of both systems, their toxicity was critically evaluated using microbial, plant, and animal models. Furthermore, comparisons with traditional Fenton (Fe2+/H2O2) and Fenton-like (Co2+/PMS) systems were made to highlight differences in modeling effects. The result revealed that solutions treated with the Fe2+/H2O2 and Co2+/PMS systems exhibited higher toxicity and a lower density of Escherichia coli (E. coli) (Supplementary Fig. 87), primarily due to the toxic effects of high concentrations of Co2+ and iron sludge on E. coli growth. In contrast, the E. coli colony density in solutions treated with PMS and PAA-activated systems was comparable to that of the control group (Fig. 7f), suggesting minimal toxicity of the degradation products. Wheat growth experiments further supported this observation, showing that the mortality rate and shoot length of wheat grown in PMS and PAA-activated system-treated solutions were comparable to those in the control group (Fig. 7d, e). In addition, zebrafish growth experiments corroborated these findings (Fig. 7g). Zebrafish cultured in the PMS and PAA-activated systems, as well as in the control group, exhibited no abnormalities or mortality during the 96 h observation period, indicating the effective detoxification capabilities of these constructed catalytic systems. In contrast, zebrafish in the Fe2+/H2O2 and Co2+/PMS systems displayed significant abnormalities, while zebrafish in the SMX control group showed mortality (Supplementary Fig. 88).
These results indicated that the PMS and PAA-activated systems using CoSAs-ZnO catalysts could effectively degrade organic pollutants while selectively generating species without introducing more toxic byproducts. This highly efficient and detoxifying integrated system demonstrates excellent potential for practical applications in water purification, highlighting its versatility and safety.
Discussion
In this study, an SAC featuring surface hydroxylation and a Co-O-Zn configuration was meticulously constructed, informed by DFT predictions. This SAC demonstrated remarkable efficacy in activating PMS and PAA, achieving high utilization rates of 86% and 89%, respectively. Notably, the degradation rate of SMX was exceptionally high in both systems, with nearly complete removal achieved within 6 min. The normalized kinetic constants for these reactions were 586.7 and 427.8 min−1 M−1, respectively, significantly surpassing previously reported values. Moreover, this SAC exhibited 96% and 95% selectivity for generating SO4•− and 1O2, respectively, in the PMS and PAA systems. Detailed DFT calculations revealed that the surface hydroxyl groups on ZnO were crucial for modulating the adsorption configurations of the two oxidants, thereby enabling the selective generation of these active species. The specificity of the active species produced in each system facilitates their application in various targeted scenarios. The SO4•−-dominated PMS activation system demonstrated high efficiency in treating electron-deficient, refractory BA wastewater, with pilot-scale experiments indicating that removal efficiency remained nearly 100% even after multiple treatments. Similarly, the 1O2-dominated PAA activation system exhibited a remarkable ability to convert benzyl alcohol to benzaldehyde, achieving an 89% selectivity. Moreover, comprehensive toxicity assessments using zebrafish, wheat, and E. coli experiments confirmed the non-toxic nature of degradation products.
This approach demonstrates the versatility of one catalyst across a range of oxidant systems and creates distinctive applications tailored to their specificities. Such versatility provides a strategy for the development of advanced SACs for both water purification and high-value chemical conversion reactions. This research establishes a foundational methodology for deploying surface-engineered SACs in environmental and chemical engineering applications, highlighting their potential to address critical needs in sustainable technology development.
Methods
Chemicals and reagents
Detailed information is provided in the Supplementary information.
Construction of Co SACs
(a) Preparation of CoSAs-ZnO. First, Zn(CH3COO)2 (2 g, 10.9 mmol) and Co(CH3COO)2 ∙ 4H2O (0.1 g, 0.4 mmol) are dissolved in 48 mL of methanol (CH3OH) and stirred for 10 min to ensure complete dissolution. Then, a pre-prepared solution (48 mL) of dimethylimidazole (4.44 g, 46.2 mmol) is poured into the zinc and cobalt acetate solution while continuous stirring is maintained for 24 h to allow for thorough mixing and reaction. Afterward, the mixture was centrifuged to separate the solid material from the liquid. The resultant precipitate was washed three times with CH3OH to remove residual impurities. The cleaned precipitate was dried under vacuum at 60 °C for 12 h to obtain a dry powder. Finally, the obtained powder was dispersed in a ceramic boat, placed in a muffle furnace, heated to 500 °C at 2 °C min−1, and maintained at this temperature for 2 h. Ultimately, a 0.062 g sample can be obtained.
(b) Preparation of ZnO. The preparation of pristine ZnO was similar to that of CoSAs-ZnO, excluding the use of Co(CH3COO)2 ∙ 4H2O. Ultimately, a 0.060 g sample can be obtained.
(c) Preparation of Co3O4. The pristine Co3O4 was prepared in a similar manner to ZnO, with the exception that 2 g (10.9 mmol) of Zn(CH3COO)2 were replaced with 2.72 g (10.9 mmol) of Co(CH3COO)2 ∙ 4H2O. Ultimately, a 0.024 g sample can be obtained.
Characterizations
X-ray absorption fine structure spectra (Co K-edge) were obtained at the BL14W1 station of the Shanghai Synchrotron Radiation Facility (SSRF). The storage rings at SSRF operated at 3.5 GeV with a peak current of 250 mA. A Si (111) monochromator was utilized to collect the data specific to the Co K-edge at room temperature. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) imaging was performed using a high-resolution FEI Titan G2 60-300 transmission electron microscope. Further characterizations of the catalysts and corresponding theoretical calculations are detailed in the Supplementary Information.
Catalytic performance evaluation
Degradation experiments were performed in a 50 mL aqueous suspension containing 0.1 g L−1 of the synthesized catalyst, 0.15 mM PMS (PMS system), 0.18 mM PAA (PAA system), and 10 mg L−1 SMX. To establish equilibrium between adsorption and desorption, the catalyst and SMX were pre-mixed and agitated for 5 min prior to the addition of PMS or PAA. Following the addition of PMS or PAA, 1 mL aliquots were sampled from the suspension every minute. These aliquots were immediately quenched with 0.1 mL of a 100 mM sodium thiosulfate solution to halt the catalytic reaction. Subsequently, the samples were centrifuged at 18630 × g for 20 min to separate suspended particulates. The supernatant was then analyzed for SMX concentration using high-performance liquid chromatography (HPLC, Agilent 1260 Infinity, Agilent Inc., USA).
DFT calculations
We carried out DFT calculations using the generalized gradient approximation (GGA) with the Perdew-Burke-Ernzerhof (PBE) formulation, as detailed in the references. Geometry optimization was performed with 2*2*1 k-points in the Brillouin zone. To represent the valence electrons, a plane-wave basis set with a kinetic energy cutoff of 400 eV was used, and the partial occupation of the Kohn-Sham orbitals was treated with a Gaussian smearing method with a width of 0.05 eV. Convergence in electronic energy calculations was achieved when the energy change was less than 10−5 eV, while geometry optimization was considered convergent if the energy change was below 0.02 eV per Ångström (eV Å−1). A 15 Å vacuum layer was added to minimize artificial interactions between periodic images. Weak interactions were handled using the DFT + D3 method and the Grimme empirical correction scheme.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.


Responses