Vacancies and sea urchin structure protect cobalt manganese spinel from anion poisoning in peroxymonosulfate activation

Introduction
Over the past years, the increasing demand for clean water has been challenged by the inevitable discharging of refractory organic pollutants from multiple industries into various aquatic waters1,2,3,4. To handle this problem, a number of water treatment technologies have been developed, among which heterogeneous peroxymonosulfate (PMS)-activation-based advanced oxidation process (PMS-AOP) appeared as a promising strategy for removal of refractory organic pollutants owing to its outstanding oxidation capability5. As well known, PMS-AOP has some obvious advantages: (i) its main reactive species sulfate radical (SO4●−) possesses a higher potential (2.5–3.1 eV) and a longer lifetime (t1/2 = 30–40 μs) than hydroxyl radical (●OH) (1.8–2.7 eV, t1/2 = 10−3 μs)6,7; (ii) PMS of solid can be easily stored and transported8; (iii) it can be operated across a wide pH range9. Further, compared with homogeneous catalytic reaction, the heterogeneous catalytic PMS-activation systems can be operated under mild conditions and easily recycled by catalyst separation10. Therefore, heterogeneous catalytic PMS-activation systems obtained much attention and many effective catalysts were developed.
To date, spinel catalysts have been widely studied for their superior catalytic properties in PMS activation due to their special AB2O4 configuration which not only guarantee high-efficiency PMS activation but also allow quick high-valency metal ion transforming to low-valency metal ion for continuously activating PMS11,12. For example, Feng et al. prepared a series of MxMn3-xO4 (M = Co, Zn, Cu, Fe; x = 1, 2) nanoparticles which showed excellent catalytic PMS activation performance for deep dechlorination (95.0%) and mineralization (65.0%) of trichloroethylene13. Xu et al. developed a series of ordered mesoporous silica SBA-15 supported bimetallic Co-Mn spinel oxides, and these materials exhibited higher PMS activation performance than monometallic catalysts14. However, the performance of most spinel catalysts in PMS-activation-based pollutant removal is unsatisfactory due to the severe interference of co-existing anions in water. According to literature, the reasons for the interference of co-existing anions include: (i) anions may react with ●OH/SO4●− free radicals to produce less reactive radicals, such as NO3−, Cl−, H2PO4−, and CO32− 15,16 (ii) anions may compete adsorption sites with PMS or pollutants, preventing PMS activation or pollutant degradation17; (iii) anions may cause catalyst agglomeration, reducing exposure of active catalytic sites on the surface of catalysts18. For example, Guo et al. prepared a series of Co3-xMnxO4 with increased Mn-O covalency, which presented efficient pollutant degradation using PMS in deionized water, but exhibited much lower pollutant degradation performance with addition of 5 mM anions19. Therefore, it is highly necessary to design a novel spinel catalyst with efficient anti-interference to anions in water when it is used to PMS-activation based pollutant degradation.
In this study, we synthesized a sea urchin-like Co-Mn spinel catalyst (CoMn2O4-S) with enriched oxygen vacancies by a facile sulfate modification strategy, which exhibited stronger resistance to anionic interference, comparing with its counterpart material, the spinel Co-Mn spinel catalyst (CoMn2O4) without sulfate modification. The sea urchin-like spinel CoMn2O4-S was systematically characterized by scanning electronic microscopy (SEM), transmission electronic microscopy (TEM), X-ray diffraction (XRD) pattern, Brunauer-Emmett-Teller (BET), Fourier transform infrared spectroscopy (FT-IR), Raman scattering spectroscopy, electronic paramagnetic resonance (EPR) spectroscopy, and X-ray photoelectron spectroscopy (XPS). Then, we studied the main active species involved in the CoMn2O4-S/PMS system and the mechanism for its enhanced anti-interference to anions. Finally, the applicability and reusability of CoMn2O4-S for PMS activation in tap and river waters were studied. This work provides a new strategy to advance the negative effects of co-existing anions on heterogeneous PMS-activation-based pollutant degradation using spinel catalysts.
Results
Characterization
We first used XRD to characterize the crystal structure of CoMn2O4-S and CoMn2O4. As shown in Fig. 1a, both the synthesized CoMn2O4-S and CoMn2O4 materials exhibited the XRD patterns of spinel crystalline structure (JCPDS 18-0410), since the diffraction peaks of the (111), (113), (311), and (004) planes matched well with spinel CoMn2O4 oxides. It should be noted that, the XRD peaks of CoMn2O4-S presented much weaker than that of CoMn2O4, suggesting that the possible formation of crystal defects by the sulfate-modification strategy. EPR spectra (Fig. 1b) further showed that CoMn2O4-S possessed a much higher symmetrical EPR peak (g = 2.003) than CoMn2O4, indicating that CoMn2O4-S contained more oxygen vacancies. In comparison, CoMn2O4 was then calcined at 450 °C for 2 h under N2 protection to obtain the CoMn2O4-control which showed no EPR peak indicating no presence of vacancies. This further confirmed the measured EPR peaks for CoMn2O4-S and CoMn2O4 were from oxygen vacancies.

a XRD spectra and b EPR spectra of CoMn2O4 and CoMn2O4-S.
We then characterized the morphological structure of the as-prepared CoMn2O4-S and CoMn2O4 by SEM. As shown in Fig. 2a, b, the CoMn2O4-S had a sea urchin-like structure with a radial distribution of nanorods, while the CoMn2O4 was composed of a bunch of stacking nanorods. It can be found that intense Co, Mn, and O signals were uniformly distributed in the energy dispersive X-ray spectroscopy (EDS) mappings of CoMn2O4-S (Supplementary Fig. 1), while a slight S signal was also observed and should be from the remanent sulfate, suggesting sulfate addition induced the formation of the sea urchin-like structure. Magnified SEM images (Fig. 2c, d) show that the average diameter sizes of the nanorods in the sea urchin-like CoMn2O4 and the CoMn2O4-S were about 85.3 nm and 186.0 nm, respectively, suggesting sulfate modification would stimulate the growth of spinel nanorod. Additionally, obvious lattice spacings of 0.224 and 0.265 nm were found in the HR-TEM images of CoMn2O4-S (Supplementary Fig. 2), which could be attributed to spinel (004) and (113) lattices19, respectively. This is consistent with the result of XRD pattern of CoMn2O4-S. Moreover, the N2 adsorption–desorption isotherms of the synthesized CoMn2O4 and CoMn2O4-S (Fig. 3a, b)) exhibited the type IV isotherm curves with an H3 hysteresis loop. The specific surface areas and the pore sizes (Supplementary Table 1) of the two catalysts are very close to each other, but the sulfate modification process slightly reduced the pore volume of the catalyst, which was probably caused by the modification process and the formation of sea urchin-like nanospheres without the mesoporous structure20.

a, c SEM images of CoMn2O4. b, d SEM images of CoMn2O4-S.
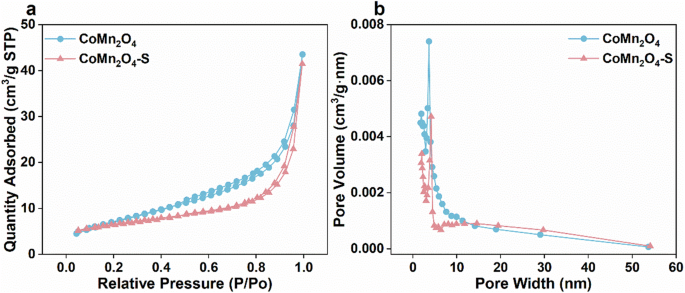
a N2 adsorption/desorption isotherms and b pore size distribution curves of CoMn2O4 and CoMn2O4-S.
We then took XPS characterization to analyze the surface elemental compositions and states of CoMn2O4 and CoMn2O4-S. The wide-scan XPS spectra in Fig. 4a show the coexistence of Mn 2p, Co 2p, S 2p, and O 1s in CoMn2O4-S and the coexistence of Co, Mn, and O in CoMn2O4. First of all, no obvious binding energy shifts were found for Co 2p and Mn 2p in CoMn2O4 and CoMn2O4-S. But the binding energy shift was observed for O 1s. The binding energy of O 1s for CoMn2O4-S was increased in comparison with CoMn2O4, which could be ascribed to an increased electron delocalization due to oxygen vacancies and possible sulfate leftover in CoMn2O4-S as probably introduced from sulfate substitution on the surface. Similar phenomena have been reported by some other research groups when modifications were introduced to supramolecule, Bi4Ti3O12, and P-CoMoO4/Co(OH)2 catalysts21,22,23. Especially for the P-CoMoO4/Co(OH)2 catalyst, the authors used a phosphorus modification strategy to remodel the surface of catalyst, which resulted in obvious oxygen vacancy formation and blue shift of O 1s binding energy.
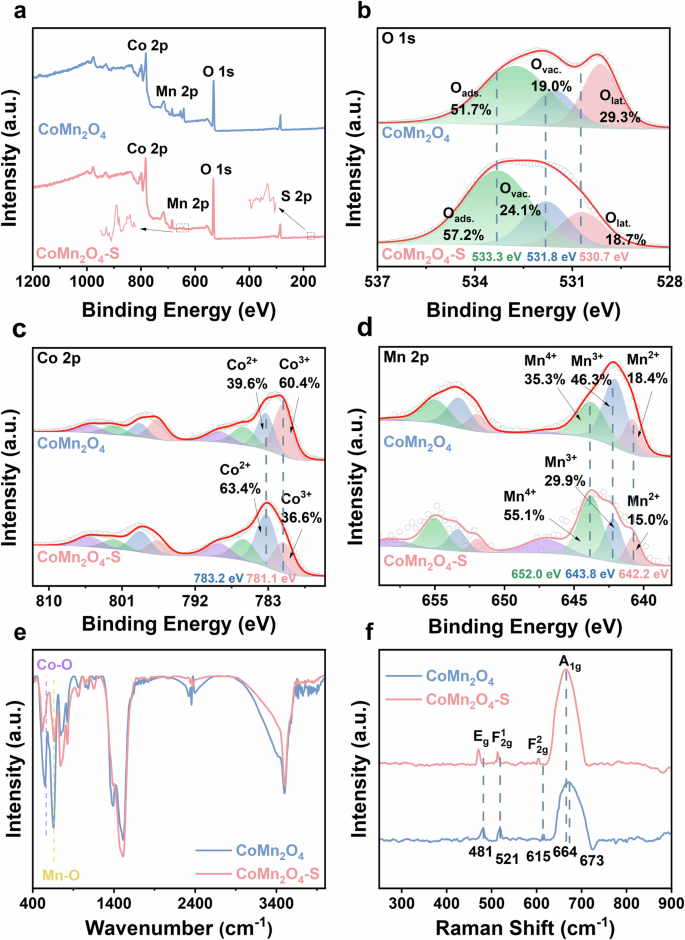
a Wide-scan XPS spectra, b High-resolution XPS spectra of O 1s, c High-resolution XPS spectra of Co 2p, d High-resolution XPS spectra of Mn 2p; e FTIR and f Raman spectra of CoMn2O4 and CoMn2O4-S.
Followed by, we started analyzing the components of those elemental XPS signals. For the O 1s spectrum of CoMn2O4, three deconvoluted peaks at 530.1, 531.5, and 532.7 eV could be attributed to lattice oxygen (Olat), oxygen vacancy (Ovac), and surface adsorbed H2O (Oads) with corresponding proportions of 29.3%, 19.0%, and 51.7%, respectively19,24. Analogously, the O 1s spectrum of CoMn2O4-S could be deconvoluted into three peaks at 530.7, 531.8, and 532.7 eV representing Olat, Ovac, and Oads with corresponding proportions of 18.7%, 24.1%, and 57.2%, respectively25. Obviously, the Ovac proportion in CoMn2O4-S is higher than that in CoMn2O4, being consistency with the EPR results (Fig. 1b). For CoMn2O4 (Fig. 4c), the Co 2p spectrum could be deconvoluted into a pair of Co3+ related peaks at Co 2p1/2 796.6 eV and Co 2p3/2 781.1 eV, a pair of Co2+ related peaks at Co 2p1/2 798.9 eV and Co 2p3/2 783.3 eV, and two pairs of shake-up satellite peaks at 786.0, 789.0, 802.0, and 805.0 eV. The proportions of Co3+ and Co2+ to Co element for CoMn2O4 were calculated as 60.4% and 39.6%, respectively26. Similarly, for the Co 2p spectrum of CoMn2O4-S (Fig. 4c), the Co 2p1/2 peak at 796.6 eV and Co 2p3/2 peak at 781.1 eV could correspond to Co3+, while the peaks of Co 2p3/2 peak at 783.2 eV and Co 2p1/2 peak at 798.7 eV could be attributed to Co2+. The calculated proportions of Co3+ and Co2+ to Co element for CoMn2O4-S were 36.6% and 63.4%, respectively. As to the Mn 2p spectrum of CoMn2O4 (Fig. 4d), the deconvoluted peaks at 640.1 and 652.0 eV could be assigned to Mn 2p3/2 and Mn 2p1/2 of Mn2+, respectively. And the peaks located at 642.1 and 653.3 eV separately correspond to Mn 2p3/2 and Mn 2p1/2 of Mn3+, while the peaks at 643.7 and 655.0 eV could be attributed to Mn 2p3/2 and Mn 2p1/2 of Mn4+, respectively12. Mn2+, Mn3+, and Mn4+ separately account for 18.4%, 46.3%, and 35.3% of the Mn composition in CoMn2O4. In regard to the Mn 2p spectrum of CoMn2O4-S (Fig. 4d), the presence of Mn2+, Mn3+, and Mn4+ could be verified due to three pairs of deconvoluted Mn 2p3/2 and Mn 2p1/2 peaks at 640.8 and 652.0 eV, 642.2 and 653.4 eV, and 643.8 and 655.0 eV, respectively. The proportions of Mn2+, Mn3+, and Mn4+ were separately calculated as 15.0%, 30.0%, and 55.0%.
The structures of CoMn2O4-S and CoMn2O4 were further analyzed by FTIR and Raman characterizations. As first shown in the FTIR spectra of CoMn2O4-S and CoMn2O4 (Fig. 4e), distinct characteristic peaks corresponding to Mn−O and Co−O stretching vibration modes were observed at 660 and 565 cm–1, respectively27. Then, Raman spectra of the prepared Co-Mn spinel catalysts in selected spectral region 900-200 cm−1 were shown in Fig. 4f. The Raman spectrum of CoMn2O4 had four well-defined peaks locating at 481, 521, 615, and 673 cm−1, agreeing with the Raman spectrum of Co3O4 spinel28,29,30. The high-frequency band at 673 cm−1, could be identified as the A1g vibration mode of octahedral sites [CoO6] in O7h symmetry. The Raman peaks at 615 cm−1, 521 cm−1, and 481 cm−1 could be assigned to F2g1, F2g2, and Eg vibration modes of tetrahedral and octahedral sites, respectively31,32,33. In comparison, CoMn2O4-S still had the four peaks of A1g F2g1, F2g2, and Eg, but all of the four peaks were shifted to lower wavenumbers, owing to the lattice defects by the occupation of the tetrahedral and octahedral sites34,35, which were probably due to the sulfate modification. Taking together the EPR (Fig. 1b), XPS (Fig. 4b), and Raman (Fig. 4f) spectra, it can be confirmed oxygen vacancies were induced by sulfate modification for bringing the lattice defects in CoMn2O4-S.
Peroxymonosulfate activation for pollutant degradation
Next, we evaluated the catalytic PMS activation performance of CoMn2O4-S and CoMn2O4 using phenol (PE) as a target pollutant first. Figure 5a shows that CoMn2O4-S possessed slightly better PMS activation performance than CoMn2O4 since the CoMn2O4-S/PMS system achieved faster PE removal kinetics with achieved a higher apparent kinetic rate constant (kapp) (0.2064 min−1) than the 0.1535 min−1 of CoMn2O4/PMS system in 18 min, although the final PE removals were both 100%. In addition, the catalytic performance of CoMn2O4-S is compared with other catalysts reported in recent literature shown in Supplementary Table 2. It should be noted that the CoMn2O4-S spinel showed the fastest and highest removal efficiencies of PE degradation (100% PE removal within 18 min). Without either catalyst or PMS, there were nearly no PE degradation (<2%) in 18 min, confirming the effectiveness of PMS activation by CoMn2O4 and CoMn2O4-S. To rule out the possible effects of leached metals on PMS activation, we tested the cobalt leaching and analyzed its direct effect on PMS activation. After CoMn2O4-S/PMS reaction for 10 and 20 min, the detected concentrations of cobalt ions were 2.46 and 3.96 mg·L−1, and no Mn leaching was detected. We then conducted phenol degradation experiments by Co2+/PMS system with using 2 and 4 mg·L−1 of Co2+ at the same reaction conditions as CoMn2O4-S/PMS system except for the addition of CoMn2O4-S. As can be seen from Supplementary Fig. 3, only 17.9% and 28.6% of phenol were degraded in 18 min by the usage of 2 and 4 mg·L−1 of Co2+, respectively, which are much lower than CoMn2O4-S/PMS system achieved (100%). Therefore, the phenol degradation in CoMn2O4-S/PMS system should be originated from the CoMn2O4-S catalytic PMS activation, instead of the leached cobalt-based homogeneous catalysis. And the catalysis of CoMn2O4-S in PMS activation could be further supported by the dose dependent PE removal efficiency of CoMn2O4-S and PMS (Supplementary Fig. 4).
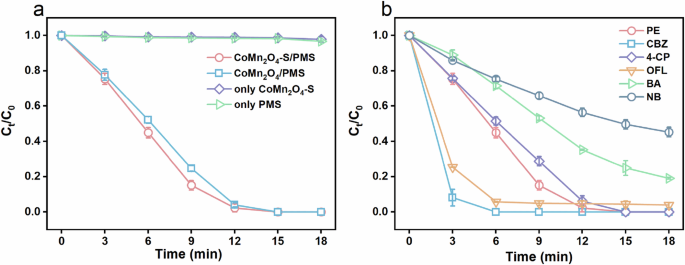
a PE degradation in different systems. b Other pollutants’ degradation in CoMn2O4-S/PMS system. Reaction conditions: [contaminant] = 7.5 mg·L−1, [PMS] = 0.4 mM, [catalyst] = 25 mg·L−1, T = 25 °C, pH0 = 3.26.
The PMS-activation performance of CoMn2O4-S was further tested using various recalcitrant pollutants, including carbamazepine (CBZ), ofloxacin (OFL), 4-chlorophenol (4-CP), benzoic acid (BA), and nitrobenzene (NB). Among them, CBZ, OFL, and 4-CP were efficiently removed by CoMn2O4-S/PMS system after 18 min reaction, while BA and NB were degraded by 81.0% and 54.8%, respectively. It is well known that BA and NB are more recalcitrant pollutants than others due to their strong electron-withdrawing groups with high antioxidation capabilities. Meanwhile, the degradation of BA and NB suggests the main active species in CoMn2O4-S/PMS system were mainly hydroxyl (●OH) or sulfate (SO4●−) radicals, since BA and NB were thought to be the probe of SO4●− and ●OH25,36. We also tested the degradation of these recalcitrant pollutants using CoMn2O4/PMS system (Supplementary Fig. 5), which could remove 100% of CBZ, 34.2% of 4-CP, 91.9% of OFL, 72.2% of BA, and 12.6% of NB within 18 min. Apparently, CoMn2O4-S/PMS outperformed CoMn2O4/PMS system over different pollutants.
Mechanism of PMS activation process
In order to identify the active species in the reaction, the radical quenching experiments were first conducted for CoMn2O4-S/PMS and CoMn2O4/PMS systems. Methanol (MeOH) was used as a scavenger for both SO4●− and ●OH due to their reaction rates with MeOH were reported to be 3.2 × 106 and 9.7 × 108 M−1 s−1, while tert-butyl alcohol (TBA) preferentially acted as a scavenger for ●OH owing to the significantly larger rate (k●OH,TBA = 6.0 × 108 M−1 s−1) than kSO4●−,TBA of 4.0 × 105 M−1 s−1 37. Figure 6a shows that the PE degradation was almost not affected with the addition of 100 mM TBA, but the PE degradation was drastically inhibited by 100 mM MeOH, implying that SO4●−, instead of ●OH, contributed to the PE degradation in the CoMn2O4/PMS system. Then, the possible contribution of 1O2 was evaluated by adding a commonly used probe L-histidine (L-His), whose second order reaction rates with SO4●−, ●OH, and 1O2 were reported to be 2.5 × 106, 5 × 109, and 1.5 × 108 M−1 s−1, respectively37. We did not use furfuryl alcohol (FFA) as the quencher of 1O2 since it was found to quench radicals as well (Supplementary Fig. 6). Owing to those similar second order reaction rates, the possible inhibition of L-His to the pollutant degradation in PMS-activation involved system is hardly to be seen as the 1O2 quenching effect. Therefore, the serious inhibition of L-His to PE degradation in CoMn2O4/PMS and CoMn2O4-S/PMS systems (Fig. 6a, b) did not absolutely indicate the main role of 1O2. To further clarify the role of the active species in the CoMn2O4/PMS and CoMn2O4-S/PMS systems, the contributions of SO4●−, ●OH, and 1O2 were calculated using Eqs. (1)−(8)37. The reaction kinetics rate constants of pollutant (e.g., PE) with ●OH, SO4●−, and 1O2 (i.e., k●OH, kSO4●−, and k1O2) were calculated via Eqs. (1)−(4). The contributions of different ROS were then evaluated via Eqs. (5)−(8). The calculated contributions of ●OH, SO4●−, and 1O2 in CoMn2O4/PMS and CoMn2O4-S/PMS systems were presented in Fig. 6c. As exhibited, the contributions of ●OH, SO4●−, and 1O2 in CoMn2O4-S/PMS system were determined as 15.3%, 73.9%, and 8.0%, respectively, while those in CoMn2O4/PMS system were separately 18.7%, 49.8%, and 28.4%. Obviously, SO4●− was the dominant reactive species for PE degradation in both of CoMn2O4/PMS and CoMn2O4-S/PMS systems, while ●OH and 1O2 made minor contributions. Additionally, it could be seen that the contribution of SO4●− in CoMn2O4-S/PMS system was higher than that in CoMn2O4/PMS system.
Where, k0 is the apparent rate constant of the control reaction without any scavenger. k1, k2, and k3 represent the apparent rate constants of PE degradation with the introduction of TBA, MeOH, and L-His, respectively. λ represents corresponding contribution of a kind of ROS.
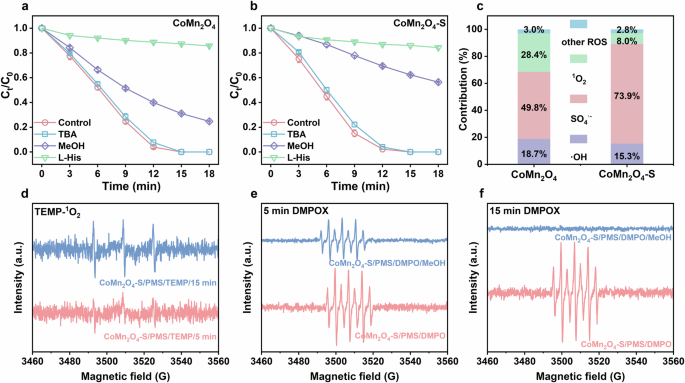
PE degradation performance in the presence of radical scavengers in a CoMn2O4/PMS and b CoMn2O4-S/PMS systems. c The contributions of ●OH, SO4●−, 1O2, and other ROS on the degradation of PE. d EPR spectra in the presence of 50 mM TEMP. EPR spectra were obtained from various systems in the presence of 50 mM DMPO after 5 min (e) and (f) 15 min. Reaction conditions: [PE]0 = 7.5 mg·L−1, [PMS]0 = 0.4 mM, [catalysts]0 = 25 mg·L−1, T = 25 °C, [TBA]0 = 100 mM, [MeOH]0 = 100 mM, [L-His]0 = 1 mM.
EPR tests were further conducted to verify the involved active species using 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) and 2,2,6,6-tetramethylpiperidinyloxyl (TEMP) as spin-trapping agents. As shown in Fig. 6d, a set of signals with three peaks with an intensity of 1:1:1 attributing to the TEMP-1O2 adduct appeared, suggesting the generation of 1O2 in CoMn2O4-S/PMS system38. The signal intensity of TEMP-1O2 increased with reaction time, illustrating the possible generation of 1O2 during the reaction process. Besides, a set of time-dependent signals with seven strong peaks of 5,5-dimethyl-2-oxo-pyrroline-1-oxyl (DMPOX) were observed in the CoMn2O4-S/PMS system (Fig. 6e, f) and could be ascribed to the oxidation of DMPO by free radicals (i.e., ●OH and SO4●−)39. The production of ●OH and SO4●− was further confirmed by the significantly decreased and nearly disappeared DMPOX peaks after the addition of MeOH for 5 min and 15 min, respectively (Fig. 6e, f). Additionally, the addition of MeOH for 15 min could nearly quench all of ●OH and SO4●− while PE degradation continued after this time (Fig. 6b), also suggesting the generation of 1O2. It should be noted that the possibility of high-valent metal-oxo generation could be ruled out for the low selectivity (52.6%) of conversion of methyl phenyl sulfoxide (PMSO) to methyl phenyl sulfone (PMSO2) (Supplementary Fig. 7). If any high-valent metal-oxo was generated due to the PMS activation, its probe PMSO would be selectively transformed to PMSO2 with an almost 100% conversion efficiency through oxygen atom transfer mechanism40. Therefore, the calculation of the contributions of ●OH, SO4●−, and 1O2 would not be influenced by any high-valent metal-oxo.
Next, we compared the XPS spectra (Supplementary Fig. 8) of fresh and used CoMn2O4-S to figure out the possible catalytic site in CoMn2O4-S for PMS activation. As shown in Supplementary Fig. 8b and Supplementary Table 3, fresh CoMn2O4-S contained 63.4% of Co (II) and 36.6% of Co (III) and the used CoMn2O4-S after PMS activation process possessed 16.2% of Co (II) and 83.8% of Co (III), presenting the PMS activation largely decreased the Co (II) content. Besides, Supplementary Fig. 8c displayed that 15.0% of Mn (II), 29.9% of Mn (III) and 55.1% of Mn (IV) were present in the fresh CoMn2O4-S. After reacting with PMS, the content of Mn (IV) reduced to 23.1%, while that of Mn (III) and Mn (II) increased to 34.6% and 23.1%, respectively. It was generally accepted that electron transfer from metal-based catalysts to PMS could efficiently generate ●OH and SO4●− and such process in turn raised up the chemical state of metal elements. In this case, the significant decrease of Co (II) and the increase of Mn (II) indicated that the Co (II) was the probable active site for PMS activation (binding energies and elemental compositions of fresh and used CoMn2O4-S presented in Supplementary Tables 4 and 5).
Based on the experimental results mentioned above, the mechanism of PMS activation by CoMn2O4-S was proposed. At first, Co (II) on the surface of CoMn2O4-S donated electrons to PMS for generate SO4●− and OH− (Supplementary Eq. (1)) and Mn (IV) and Mn (III) abstracted electrons from PMS for generating SO5●− (Supplementary Eqs. (2) and (3)) which could be used for the production of 1O2 (Supplementary Eq. (4))19. ●OH could be generated through the reaction between SO4●− and H2O/OH− (Supplementary Eqs. (5) and (6))25. Finally, SO4●−, ●OH, and 1O2 worked together to degrade pollutants (Supplementary Eq. (7)).
Enhanced anti-interference to anion poisoning
Ubiquitous anions in water (NO3−, Cl−, CO32−, SO42− and H2PO4−) may interfere with PMS-activation process; therefore, their effects on PE degradation in CoMn2O4/PMS and CoMn2O4-S/PMS systems were investigated in detail (Fig. 7). Figure 7a shows that NO3−, Cl−, SO42−, and H2PO4− presented obvious inhibition to PE degradation by CoMn2O4, but CO32− promoted PE degradation. The corresponding kapp of PE degradation with the presence of NO3−, Cl−, SO42−, and H2PO4− were 0.0557, 0.0649, 0.0698, and 0.1251 min−1, respectively, which were separately 63.7%, 57.7%, 54.5%, and 18.5% lower than the CoMn2O4/PMS system without any anion (0.1535 min−1). In comparison, the kapp of PE degradation by CoMn2O4-S with the presence of NO3−, Cl−, SO42−, and H2PO4− were 0.0885, 0.1544, 0.1564, and 0.5164 min−1, respectively. Interestingly, the inhibition rates of NO3−, Cl−, and SO42− to kapp in CoMn2O4-S/PMS system separately decreased to 57.1%, 25.2%, and 24.2%, while H2PO4− significantly increased the kapp by 150.2%.
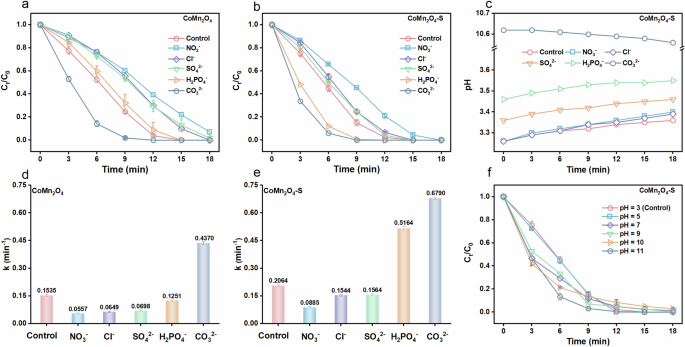
Effect of anions on degradation of PE in a CoMn2O4/PMS and b CoMn2O4-S/PMS systems. c The pH variations in the CoMn2O4-S/PMS system with different anions. The corresponding kinetic rate constants in d CoMn2O4/PMS and e CoMn2O4-S/PMS systems with different anions. f Effect of initial pH on the degradation of PE in CoMn2O4-S/PMS system. Reaction conditions: [PE] = 7.5 mg·L−1, [PMS] = 0.4 mM, [catalysts] = 25 mg·L−1, [anions] = 5 mM, T = 25 °C.
It is meaningful to weaken the inhibition of co-existing anions to pollutant degradation by advanced oxidation processes. Therefore, we further conducted some experiments to clarify the mechanism for the lowered inhibition rates of NO3−, Cl−, and SO42−, and the strengthened degradation performance by H2PO4−. At first, we monitored the effects of NO3−, Cl−, SO42− and H2PO4− on the pH in CoMn2O4-S/PMS system. As shown in Fig. 7c, the pH in the pristine CoMn2O4-S/PMS system slightly increased from 3.26 at 0 min to 3.35 at 18 min, e.g. 0.09 units of pH increase. It was found that the addition of 5 mM NO3− and Cl− resulted in a larger increase (0.13 units) of pH over 18-min reaction, while SO42− and H2PO4− raised the pH by 0.1–0.2 units at 0 min but kept pHs below ~3.5 during the 18-min reaction. And the addition of 5 mM CO32− drastically elevated the pH to 10.61 which underwent a faint decrease to 10.55 in 18 min. First, the pH changes by the addition of anions bring an interference in judging the effects of anions. Therefore, we also studied the effects of initial pH on PE degradation in the CoMn2O4-S/PMS system. As shown in Fig. 7f, increase of pH from 3 to 10 exhibited a slight promotion to PE degradation, but further increasing pH to 11 provided a more obvious promotion. We then made a comparison between anion and pH effects to give a clearer analysis, as shown in Supplementary Fig. 9. The kapp at pH 3–10 were just 1.0–1.4 times of that of the control whose pH was naturally 3.26 without adjusting pH. Apparently, only if anion effects fully suppress the pH effect, they will get a chance to be statistically significant to observe. Fortunately, the anions, paid attention in this study, all made more obvious influences on PE degradation than pH could lead to. For example, the pHs for NO3−, Cl−, SO42−, and H2PO4− added were 3.20–3.55 located in the range of pH 3–5, which gave a neglectable impact on phenol degradation rate (Supplementary Fig. 9a). In contrast, Supplementary Fig. 9b shows that obvious drops of kapp were found for NO3−, Cl−, and SO42−, and a much more significant change of increase was observed for H2PO4−. This made us have the possibility to judge whether it’s an inhibitory or promotional effect. Similarly, the pH for CO32− added was ~10.6 which is close to pH 11, at which a kapp of 1.9 times of the control was obtained. But the kapp for CO32− addition was 3.3-fold of that of control, suggesting the promotional effect of CO32−. Concludingly, the influences of anions should be from their own instead of the resulting pH changes. Therefore, the negative effects of NO3−, Cl−, and SO42− and the positive effects of H2PO4− and CO32− for CoMn2O4-S/PMS system could be confirmed.
Since CO32− presented consistent promotion to CoMn2O4/PMS and CoMn2O4-S/PMS systems, we paid attention to its mechanism accordingly. The promotional effect of CO32− could be explained by two aspects: (1) the significantly elevated pH improved PE degradation efficiency and (2) CO32− reacted with ●OH/SO4●− to form CO3●− (Supplementary Eqs. (12)−(14)) possessing high reaction activity with phenolic pollutants37. Through these discussions, it can be found that the pH variations by anions were able to illustrate the promotion of CO32− both in CoMn2O4/PMS and CoMn2O4-S/PMS systems, but not efficiently interpreting the effects of NO3−, Cl−, SO42−, and H2PO4−. Consequently, some other more information needs to be taken into consideration.
Generally, the inhibitory effects of anions could be ascribed to the competition of adsorption sites and quenching of ●OH/SO4●−. As ●OH/SO4●− were the main active species in both the CoMn2O4/PMS and CoMn2O4-S/PMS systems, the different inhibitory rates of PE degradation by anions might be attributing to the changed competition of adsorption sites41. Then, we measured zeta potentials of CoMn2O4 and CoMn2O4-S in water with different pHs. As shown in the Supplementary Fig. 10, the zeta potentials of CoMn2O4 at different pHs were very similar with those of CoMn2O4-S. The zeta potentials of CoMn2O4 and CoMn2O4-S gradually decreased from around 35 mV at pH 3 to zero at pH 6.5 and to around −25 mV at pH 9. As discussed before, the addition of 5 mM NO3−, Cl−, SO42−, and H2PO4− would slightly elevate the pH during reactions for CoMn2O4-S/PMS system (but still under acidic conditions), thus could lead to a decrease of zeta potential. Normally, decrease of absolute zeta potential would weaken the electrostatic repulsion among the solids in water, consequently spurring the agglomeration of suspended solids and causing the abatement of exposed active sites in catalysts42, which would be the case for both CoMn2O4 facing an increase of pH by the addition of 5 mM NO3−, Cl−, SO42−, and H2PO4−. We analyzed the size distribution of CoMn2O4 and CoMn2O4-S in different salt solutions (Supplementary Fig. 11). As shown, the D90 of CoMn2O4 increased sharply in the tested salt solutions of Cl−, SO42−, H2PO4−, and CO32−, while the D90 of CoMn2O4-S all got a slightly change in all of the salt solutions. Especially, CoMn2O4-S even nearly did not become any bigger in the presence of H2PO4− and CO32−. In addition, the size of CoMn2O4 (~1500 nm) in deionized water is much larger than that of CoMn2O4-S (~750 nm). Therefore, it could be demonstrated CoMn2O4 would bear much more obvious aggregation than CoMn2O4-S does in solutions. It means that, in comparison with CoMn2O4 of stacking nanorods, CoMn2O4-S would not have a big problem in agglomeration due to the sea urchin-like structure would inhibit the serious face-to-face stacking of nanobelts or nanorods. Under this circumstance, the reduction of active sites in CoMn2O4-S would be much less than that in CoMn2O4. Hence, the inhibitory rates of NO3−, Cl−, and SO42− to PE degradation by CoMn2O4-S could be obviously lower than those by CoMn2O4. But this is insufficient to explain that the effect of H2PO4− on PE degradation was switched from an inhibitory feature in CoMn2O4/PMS system to a promotional characteristic in CoMn2O4-S/PMS system. It was reported that H2PO4− can quench SO4●− and ●OH, thus inhibiting the degradation of pollutants (Supplementary Eq. (11))43. This could be the reason why the addition of H2PO4− inhibited the reaction in the CoMn2O4/PMS system, but also is hard to demonstrate the promotional effect in CoMn2O4-S/PMS system. Interestingly, a previous study reported that oxygen anions can modulate solid-liquid interfaces for accelerating surface remodeling which can promote oxygen-containing species transferring from the modulated solid-liquid surfaces to bulk of liquid44. Also, surface remodeling was reported to be enhanced by oxygen vacancy and thus could change the interaction between catalysts and reactants, influence adsorption, dissociation, and diffusion processes, finally improving catalytic activity45. XPS results (Supplementary Figs. 8 and 12) show that the chemical environment in CoMn2O4-S obtained greater changes after PMS activation than CoMn2O4, reflecting the remodeling of catalyst surface in CoMn2O4-S was much more significant than that in CoMn2O4. To further confirm the surface remodeling, we conducted XPS analyses for CoMn2O4 and CoMn2O4-S samples after reaction with PMS in the presence of H2PO4−. As shown in Supplementary Fig. 13, P−O chemical bond was observed for both CoMn2O4 and CoMn2O4-S after H2PO4− contained PMS activation reactions, indicating H2PO4− really was involved in such PMS activations. The sulfate in CoMn2O4-S was preserved after reaction, marking its role in holding oxygen vacancies. Additionally, the elemental compositions of Co 2p, Mn 2p, and O 1s were all preserved but differently shifted for CoMn2O4 and CoMn2O4-S after reaction with H2PO4−. We made comparisons of binding energy shift for CoMn2O4 and CoMn2O4-S in Supplementary Tables 6 and 7 for a better exhibition. Significantly, CoMn2O4-S underwent more obvious binding energy shifts than for CoMn2O4 did Co 2p, Mn 2p, and O 1s. For example, the shifts of Co 2p3/2 (III) and Co 2p1/2 (II) were 1.0 and −0.5 eV respectively for CoMn2O4-S, which were more significant than that (0.7 and 0 eV) for CoMn2O4. Additionally, the shifts of Mn 2p (II) and Mn 2p (IV) were 0.7 and 1.5 eV respectively, which were higher than that (0.5 and 0.9 eV) for CoMn2O4. The same is for comparing Olat 1s shifts of CoMn2O4-S (−1.3 eV) vs. CoMn2O4 (−0.4 eV). These results illustrate H2PO4− could be involved in the surface remodeling of CoMn2O4-S and CoMn2O4 during their reactions with PMS, and could help more with CoMn2O4-S. Consequently, oxygen vacancies in CoMn2O4-S and H2PO4− could synergistically improve PMS activation performance of spinel catalyst through the enhanced surface remodeling, and the resulting accelerated transfer of ●OH/SO4●− to bulk of liquid where pollutants could be easily found and reacted with.
Furthermore, linear sweep voltammetry (LSV) experiments were used to study the effect of H2PO4− on PMS activation process. LSV curves were measured for CoMn2O4/PMS and CoMn2O4-S/PMS systems with the absence and presence of PE and H2PO4−. As seen from Fig. 8a, b, PE addition decreased current intensity for both of CoMn2O4/PMS and CoMn2O4-S/PMS systems, suggesting PE consumed PMS and led to less electrons transferring from spinel catalysts to PMS electrolyte46,47. For CoMn2O4/PMS system, further addition of H2PO4− tremendously raised current intensity, which could be ascribed to the enhanced conductivity for the increased concentration of ions and the possible reverse electron transfer from PMS to CoMn2O4 electrode (unbeneficial to radical generations in PMS activation). However, in the CoMn2O4-S/PMS system, the addition of H2PO4− further decreased current intensity, which means the electron transfer from catalyst electrode to PMS was strengthened by H2PO4−. Therefore, we could deduce that H2PO4− promoted the diffusion of oxygen-containing species at the solid-liquid interface into bulk liquid, thereby promoting the collision between ●OH/SO4●− and PE for PE degradation (Fig. 8c). In summary, the unique sea urchin-like structure and enriched oxygen vacancies were the main reasons for the enhanced anti-interference of CoMn2O4-S to anion poisoning.
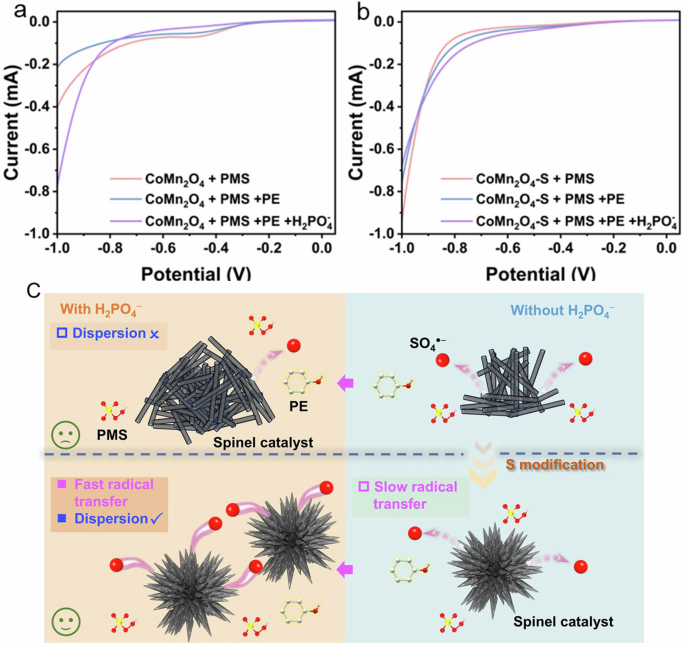
LSV curves of a CoMn2O4 and b CoMn2O4-S. c Possible degradation mechanisms for the promotional effect of phosphate in CoMn2O4-S/PMS system.
Applicability and reusability
The applicability and reusability of CoMn2O4-S were tested using tap water and river water (Fig. 9). For a comparison, we also measured the catalytic performance of CoMn2O4 in tap and river waters. As shown in Fig. 9a, tap and river waters decreased PE degradation efficiency (18 min) of CoMn2O4/PMS system from 100% to 94.3% and 78.7%, respectively. In contrast, CoMn2O4-S/PMS system (Fig. 9b) maintained 93.7% and 95.1% of PE removal in tap and river waters, respectively. Additionally, the excellent reusability of CoMn2O4-S could be illustrated by the almost no decrease of PE removal efficiency after 4 cycles of running in tap and river waters (Fig. 9c and d), which can be further confirmed in deionized water system (Supplementary Fig. 14).
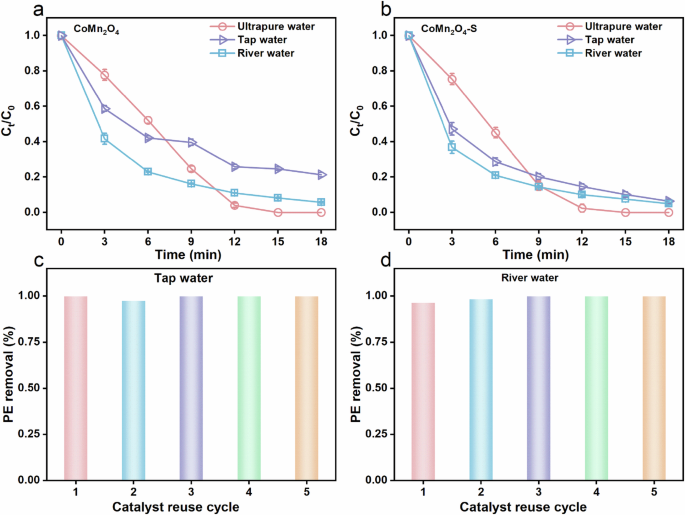
Effect of water matrix on the degradation of PE in a CoMn2O4/PMS and b CoMn2O4-S/PMS systems. Successive tests of CoMn2O4-S in c tap water and in d river water. Reaction conditions: [PE] = 7.5 mg·L−1, [PMS] = 0.4 mM, [catalysts] = 25 mg·L−1, T = 25 °C.
Discussion
In summary, a sea urchin-like CoMn2O4-S spinel catalyst with abundant oxygen vacancies was developed to present an excellent catalytic performance for PMS activation, especially in the presence of co-existing anions. First, the enhanced anti-interference to anion poisoning originated from the unique sea urchin-like structure of CoMn2O4-S. Co-existing NO3−, Cl−, and SO42− were found to decrease the zeta potential of CoMn2O4-S, and could trigger catalyst agglomeration and abatement of active catalytic sites. However, the unique sea urchin-like structure of CoMn2O4-S could weaken the catalyst agglomeration-induced abatement of reactive sites, thereby reducing the inhibitory effect of NO3−, Cl−, and SO42− to PE degradation in CoMn2O4-S/PMS system. Second, the enriched oxygen vacancies in CoMn2O4-S synergistically coupled with H2PO4− for promoting mainlySO4●− diffusing from catalyst surface to bulk liquid, consequently enhancing PE degradation in CoMn2O4-S/PMS system. Additionally, CoMn2O4-S presented excellent PMS-activation performance in tap and river waters for five cycles of running. This study provides a feasible strategy for enhancing the PMS-activation based pollutant degradation in the presence of co-existing anions.
Methods
Chemicals
Urea ((NH2)2CO), manganese chloride tetrahydrate (MnCl2·4H2O), tert-butyl alcohol (TBA), L-Histidine (L-His), methanol (MeOH), 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) and 2,2,6,6-tetramethylpiperidinyloxyl (TEMP) were purchased from Shanghai Macklin Biochemical Co., Ltd (China). Peroxymonosulfate (PMS), phenol (PE), ammonium fluoride (NH4F), cobalt nitrate hexahydrate (Co(NO3)2·6H2O), sodium sulfate anhydrous (Na2SO4), nitrobenzene (NB), benzoic acid (BA), 4-chlorophenol (4-CP), ofloxacin (OFL), carbamazepine (CBZ), and methyl phenyl sulfoxide (PMSO) were purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. (China). All the chemicals were of analytical grade and were used without further treatment unless otherwise specified.
Preparation of spinel catalysts
The CoMn2O4-S catalysts were synthesized via the following steps as illustrated in Scheme 1. At first, a cobalt oxide precursor (Co(CO3)0.5(OH)·0.11H2O (CoCH)) was synthesized by a modified hydrothermal method.0.665 g of Co(NO3)2·6H2O, 0.686 g of (NH2)2CO, and 0.254 g of NH4F were dissolved in 80 mL of deionized water to form a homogeneous solution, which was then transferred into a 100 mL teflon-lined stainless steel autoclave for 5 h of hydrothermal treatment at 393 K48. After cooling down to room temperature (approximately 298 K), the products were separated by centrifugation, washed with deionized water and ethanol for three times, and dried before using. hydrothermal method. Then, CoCH (0.2 g), MnCl2·4H2O (0.7 g), and Na2SO4 (2.1 g) were dissolved in 250 mL of ultrapure water. This solution was heated at 393 K for 1 h under stirring for producing some solid products which were then centrifuged and washed with deionized water and ethanol each for three times. After drying at 333 K for 12 h, these solid products were annealed in air at 473 K for 3 h with a heating rate of 3 K·min−1. After cooling down to room temperature, the expected vacancy-containing sea urchin-like spinel CoMn2O4-S was obtained the usage of Na2SO419.
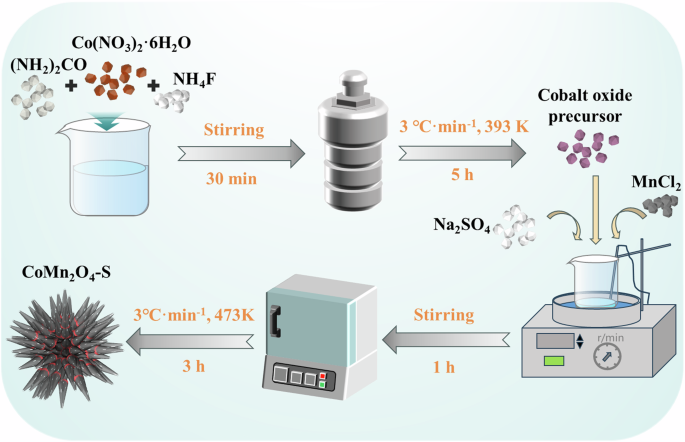
Prepare process of CoMn2O4-S.
Pollutant degradation by PMS activation
The PMS-activation based pollutant degradation experiments were carried out in 100 mL glass beakers containing 7.5 mg·L−1 of PE and certain amounts of PMS and spinel catalysts at room temperature (298 ± 2 K). For PE analysis, 1.0 mL of the reaction suspension was collected at certain time intervals (0, 3, 6, 9, 12, 15, and 18 min), then quenched rapidly with 1.0 mL of MeOH, and filtrated by 0.22 μm cellulose acetate syringe membrane filters. The main reactive oxygen species (ROS) for pollutant degradation were analyzed via adding different quenching agents (MeOH, TBA, and L-His). The effects of water quality parameters on pollutants degradation were studied by adding different concentrations of NO3−, Cl−, CO32−, SO42−, and H2PO4− and by adjusting pH to a desired value. All the experiments were carried out in duplicate or triplicate.
Characterization
The morphological structure of the samples was characterized by scanning electron microscopy (SEM, ZEISS Sigma 300) and transmission electron microscopy (TEM, Talos F200X, USA). The X-ray diffraction patterns of CoMn2O4 and CoMn2O4-S were measured by an X-ray powder diffractometer (XRD, Bruker D8 Advance, Germany). Surface elemental compositions and species were analyzed using an X-ray photoelectron spectroscopy (XPS, Thermo, USA) equipped with Al Kα radiation and the binding energy was calibrated by the standard C 1s peak at 284.8 eV. The specific surface area and pore size distribution was calculated from N2 adsorption/desorption isotherm using the Brunauer-Emmett-Teller (BET) and Barrett-Joyner-Halenda theory (BJH) models. Chemical environments of CoMn2O4 and CoMn2O4-S were measured on a Fourier transform infrared spectroscopy (FT-IR, Nicolet 5700, Thermo Electron Scientific Instrument Co. U.S.A). Raman spectra were measured using WiTech alpha300R Raman Spectroscopy equipped with a green laser of 780 nm. Zeta potentials were measured on a Zetasizer Nano-ZS90 (Malvern, USA). A pH meter (FE20, Mettler toledo, China) was used to measure pH values in solution. A CHI660E electrochemical workstation was used to characterize the relevant electrochemical properties, which was equipped with a standard three-electrode system consisting of a saturated Ag/AgCl electrode as the reference electrode, a platinum wire as the counter electrode and a catalyst-coated FTO glass as the working electrode.
Analytical methods
The concentrations of PE, NB, BA, 4-CP, OFL, and CBZ were measured using a high-performance liquid chromatography (HPLC) system (Shimadzu, Japan) with a PerkinElmer C18 reverse-phase column (250 × 4.6 mm, 5 μm particle size). Detailed mobile phase composition and detection wavelength were shown in Table 1. The zeta potential and size distribution of materials in different conditions were measured by Malvern Zetasizer Nano ZS90 and Mastersizer 3000, respectively. Electron paramagnetic resonance spectroscopy (EPR, Bruker EMX plus, Germany) was performed to detect free radicals and singlet oxygen. The DMPO and TEMP were employed as the spin-trapping agent to capture ●OH/SO4●− and 1O2, respectively.
PE removal (%) was calculated via Eq. (9).
where C0 and Ct is the concentration of PE at start time and reaction time (t), respectively.
The PE degradation kinetics were modeled by pseudo-first-order rate equation Eq. (10).
where k is the pseudo-first-order rate constant (min−1).

Responses